Screening of potential active compounds in Eurycoma longifolia against hepatic carcinoma by Network pharmacology
2018-07-05ChunxinZouWenyuZhoZhiyngYnXioxioHungShojingSongXioboWng
Chunxin Zou, Wenyu Zho, Zhiyng Yn, Xioxio Hung,b*, Shojing Song, Xiobo Wng,b*
a Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang 110016, China;
b Chinese People’s Liberation Army 210 Hospital, Dalian 116021, China
1 Introduction
Liver cancer (also known as hepatocarcinoma)is a common carcinoma, and its fatality rate is high,almost all patients died within a year [1]. About 128 anti-cancer drugs are currently derived from natural products [2]. But it still needs to find newer anti-cancer drugs because of the side effects and drug resistance of cancer therapies. Several studies reported that E.longifolia has the potential to treat cancer due to its active ingredients [3-5]. Thus, the liver cancer target proteins were used in this study.
E. Longifolia, a tropical plant, is widely used in Southeast Asia, such as Malaysia, Indonesia and it is also known as “Malaysian ginseng” because of its significant effect in health protection [6]. Its medicinal parts include roots, stems and leaves,but the roots are foremost. From the roots, several kinds of compounds have been isolated including quassinoids, canthin-6-one alkaloids, β-carboline alkaloids, tirucallane-type triterpenes, squalene derivatives, and biphenyl neolignans. Pharmacological study reported that some of these constituents were found to have anti-tumor, antimalarial, antipyretic,antiulcer, and other activities [7-11]. Therefore, 110 compounds mainly embracing quassinoids, alkaloids,squalene derivatives and biphenyl neolignans in E.Longifolia were selected as the database to research the anti-cancer network pharmacology. Despite anticancer activity of E. Longifolia which has been reported for many times, its systems-level mechanism research of compounds with liver-cancer target proteins is insufficient. Razak [12] et al. reported that the extract of E. longifolia was found to be cytotoxic against Hep2 and HFL1 cell lines, with IC50of 11 µg/mL and 13 µg/mL, respectively.
It is crucial to identify the drug-target interactions for the better understanding to the mechanism of action of a compound at the molecular level [13]. However, experimentally screen of all possible active compounds, proteins and interactions between proteins and compounds are difficult because the number of pairwise interactions is too large [14].Thus it is necessary to find a new way to carry out a specific study. Recently, network pharmacology analysis plays an important role to identify the potential drug targets and potential active ingredients.It refers to integrate the network of drug interactions and biological networks together, analysis the drug’s interaction with specific nodes in this network to understand the interactive relationships of drugs [15].These theoretical methods are not only faster but also can accurately discriminate potential drug-target interactions with the aim of providing supporting evidence for experimental studies [16]. Herein,based on the network pharmacology, we analyzed the potential active ingredients and potential target proteins. This study provides a better idea for the treatment of liver cancer.
2 Material and methods
2.1 Establish compounds database
According to the available literature, a total of 110 compounds including quassinoids, alkaloids, and other chemical constituents were collected. As far as we know, it contains almost all of the structures currently isolated from E. Longifolia. We use “DK”,“DS”, “DQ” to represent the quassinoids,alkaloids and other components respectively.Then, optimize the compounds structure using the Chembiodraw and Sybyl-X (version 2.0, TRIPOS Inc.)software, which include Structural optimization plugin.
2.2 Selecting and disposing of target protein
As we use the key words of liver cancer to search, 48 target proteins were screened according to the existence of protein-ligand complex, crystal structure determination method and higher resolution from the Therapeutic Target database (TTD; http://bidd.nus.edu.sg/group/cjttd/) and the Drug Bank database (http://www.drugbank.ca/). Then we obtain the crystal structure of the target protein complex containing the original ligand from the RCSB Protein Data Bank database (PDB; http://www.rcsb.org/pdb/home/home.do). Finally, target proteins were modified with Sybyl-X (version 2.0, TRIPOS, Inc.)software to remove the original ligand, hydrogenate,remove water, optimize and repair amino acid residues.
2.3 Molecular docking
Molecular docking technology as an important means for virtual screening is based on the known protein and small molecule compounds to imitate their geometric matching and energy matching to identify each other. This experiment use the Surflex-Dock plug-in belonging to Sybyl-X (version 2.0, TRIPOS,Inc.) software for molecular semi- flexible docking. In the first place, Sybyl-X (version 2.0, TRIPOS, Inc.) was used to set up a molecular form for the compounds to be sorted out. Then we import molecular forms and proteins into Sybyl-X (version 2.0, TRIPOS, Inc.)respectively for molecular docking. Finally, we choose the molecules with the docking score higher than 7.0.
2.4 Networks construction
The compound-cancer target network was established by the plug-in network analyzer in Cytoscape 3.2.1. In the resulting network, compounds and target proteins were represented by nodes, the edges represented various associations between the compound and the target protein when they interact with each other. The key parameters include degree of network, betweenness and so on. The active groups and the key targets were predicted from the obtained parameters.
2.5 Prediction of the ligand-binding mechanisms
Based on the above docking results, in order to further determine their ligation patterns, we used iGEM Dock 2.1 to connect these 31 compounds with Nuclear receptor coactivator 2 (3up3) which may be the key protein according to previous experiment.Then, eight compounds with good docking results were selected and analyzed binding mode by DS v4.5 (Discovery Studio visualizer). Pharmacological interactions are used in the software to understand the ligand-binding mechanisms of proteins and phytochemicals.
3 Result and discussion
3.1 Prediction of the active compounds and potential targets
This network included the information of compounds, targets and the interactions between them. The result showed that 31 compounds have high binding affinity, when compared with the original ligand along with other compounds in the database, as shown in Fig. 1. In addition, five target proteins including nuclear receptor coactivator 2, glutamine synthetase, histone deacetylase 4,heat shock protein 90, GMP Synthase [glutaminehydrolyzing] are more closely associated with the compounds and nuclear receptor coactivator 2 showed the most, according to the parameters exhibited in Table 1. “Betweenness Centrality”,“Closeness Centrality” and “Degree” are important parameters in network pharmacology.Degree and betweenness centrality stand for the influence level of the compound or target protein on the entire network, for example, the closer two ligands are in a drug-target network, the larger the value is.
3.2 Elucidation of Promising Compounds
More than 100 compounds have been isolated from E. Longifolia, they are quassinoids, canthin-6-one alkaloids, β-carboline alkaloids, tirucallane type triterpenes, squalene derivatives, biphenyl neolignan,and other components [17]. We compared the 31 compounds with the score greater than seven, DQ-9,DQ-10, DQ-11, DQ-12, DQ-13, DQ-14, DQ-15 and DS-17 showed more potent activities than the others.Thus, these eight candidate compounds were identified as main effective components. The structures are listed in Fig. 2.
Four squalene derivatives were demonstrated to have good activity in network pharmacology, which are DQ-9, DQ-10, DQ-11, DQ-12. They all characterized as eight asymmetric carbons and two or three tetrahydrofumn rings. Morita H [18] et al researched the relationship of the structure and conformation of the compound with their cytotoxicity on KB cells which showed all of them have cytotoxicity and 14-deacetyl eurylene (DQ-10) showed the most potent activity. Our research showed that such ingredients may also have good anti-liver cancer activity.
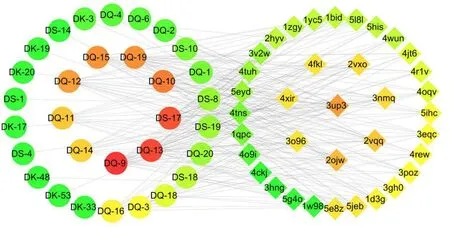
Fig. 1 Main effective component-target network.Different shapes represent the nodes with different attributions. Circle nodes represent the main effective components; diamond stand for the key target. The degree of affinity is proportional to the color from red, yellow to green
Biphenyl neolignan is a class of active ingredients in E. longifolia. Morita H [19] et al isolated four novel compounds including two biphenyls and two diphenyl ethers from the wood of E. Longifolia. The number of target proteins directly interacting with DQ-13, DQ-14, DQ-15 is greater than 10.
Alkaloids with dynamic pharmacological properties is one of the major constituents in E.longifolia. Types among them mostly are canthin-6-one, canthin-5,6-dione and β-carboline alkaloids.DS-17 belongs to β-carboline alkaloids. Kuo PC [3] et al.isolated three new alkaloids from E. longifolia incluing DS-17, 5-hydroxymethyl-9-methoxycanthin-6-one,1-hydroxy-9-methoxycanthin-6-one and tested their in-vitro cytotoxic and antimalarial activities. DS-17 did not show significant cytotoxicity against human lung cancer (A-549) and human breast cancer (MCF-7) cell lines compared with others. However, its anti-liver cancer activity needs further study because on this network it forms interactions with the 23 liver cancer protein targets.

Table 1 The parameters of the most promising compounds and targets
3.3 Prediction of binding modes
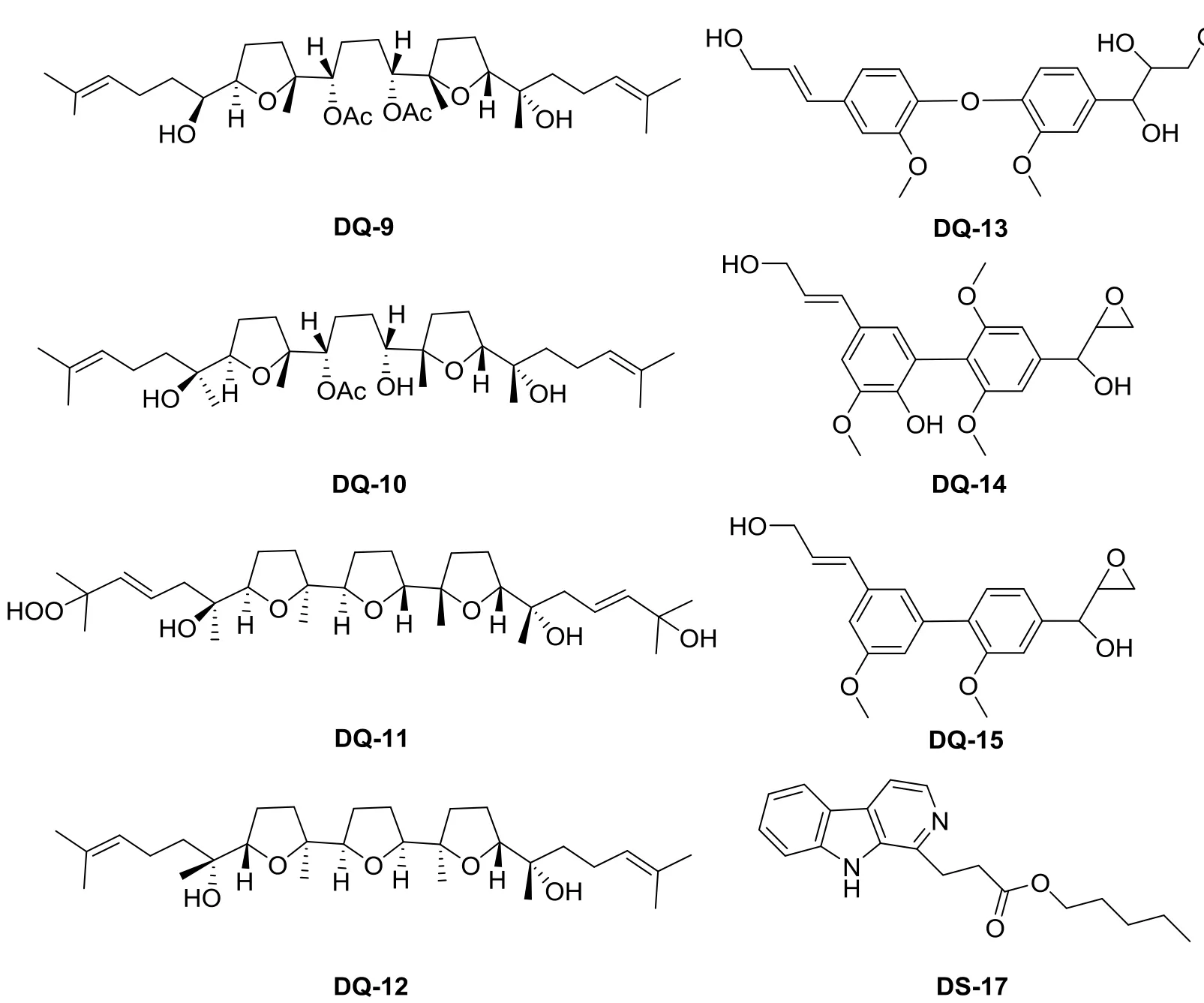
Fig. 2 Eight candidate compounds from E. longifolia
For the further research on the binding modes of the eight compounds, nuclear receptor coactivator 2 protein was employed in the process of docking,which have the highest degree values based on the target-cancer network. Nuclear receptor coactivator 2 (3up3), as a coactivator, plays an important role in androgen receptor of cancer cells when treated with antiandrogens [20]. We used iGEMDock 2.1 to connect these eight compounds with 3up3 protein,respectively. The interactions of these ligands with modelled protein were revealed according to Total energy, VDW (van der Waal energy) and H-Bond(Hydrogen bond) interaction [21]. H-bond and Elec terms are hydrogen bonding energy and electro statistic energy, respectively. The resulting profiles of interactions and analysis results were shown in Table 2. The leading compound was believed to have smaller dissociation constant and higher binding energy, and VDW with receptor. Therefore squalene derivatives and biphenyl neolignan could be considered as lead molecule in designing new formulation of drug in targeting liver cancer.
All selected compounds showed high binding affinity with the targets. As shown in Fig. 4, we found that all of them were bound at the active site of the 3up3 target structure. In order to further observe the connection of compounds and proteins, we selected the most active compounds in each classes, which isDQ-9 for squalene derivatives, DQ-13 for biphenyl neolignans, DS-17 for alkaloids to show their closeup view. As exhibited in Fig. 5, compound DQ-9 formed six pi-alkyl interactions with ARG 532, ARG 536, PHE 478 and TRP 544, ILE 657, PHE 560 residues. In addition, one hydrogen bond interaction was observed between the hydrogen on oxygen atom in compound DQ-9 and THR 530 residues.Compound DQ-13 formed two pi-pi stacked and three hydrogen bond interactions with amino acid residues. Additionally, DS-17 mainly interact with the amino acids residues through the van der Waals.
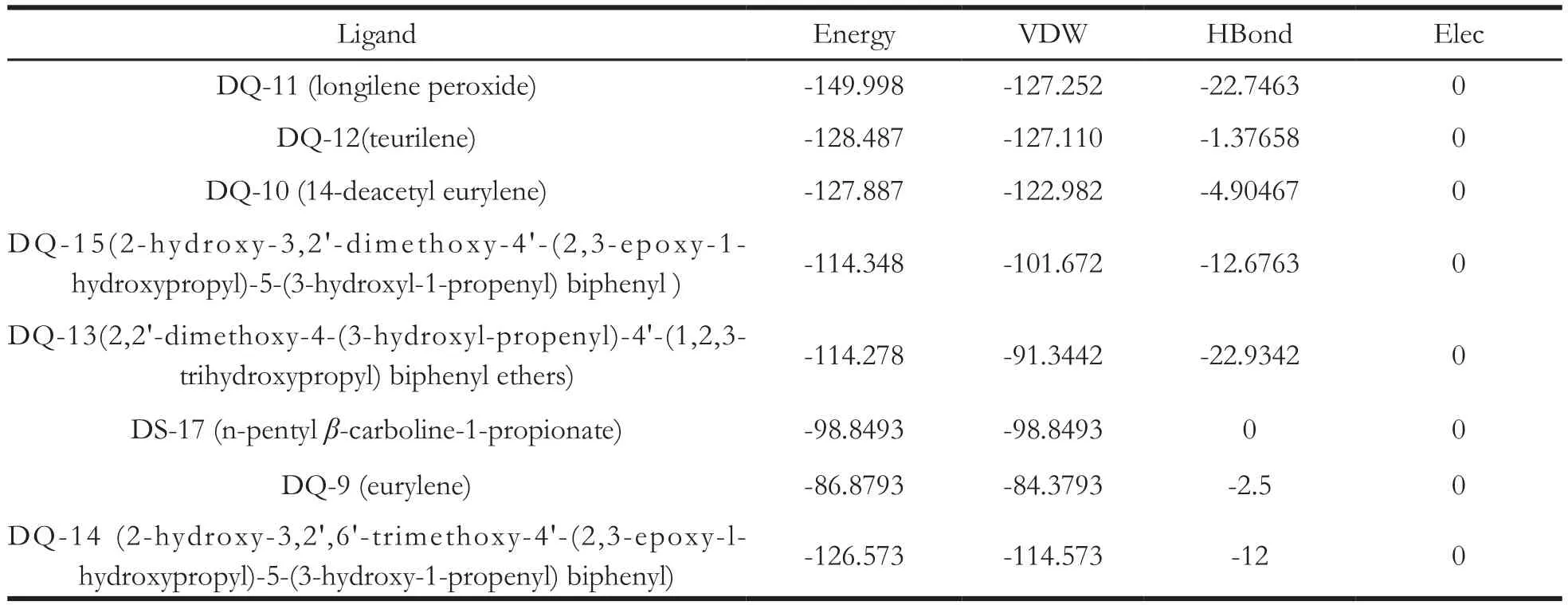
Table 2 The interaction energies (kcal·mol-1) of 3up3 and 8 plant compounds obtained from the molecular docking with iGEM Dock
According to the mode of action, since squalene derivatives contain a longer chain and multiple oxygen atoms in their structure, so van der Waal energy and hydrogen bonds formed with GLY, LEU, and THR residues are of great importance. Biphenyl neolignans mainly form van der Waal energy through interaction with many amino acids residues, and its phenyl with PHE, TRP form pi-pi stacked. In addition, these alkaloids also interact with a variety of amino acids residues in both ways, which are balanced.
4 Conclusion
In conclusion, 110 compounds from database were docked with 48 liver cancer proteins. 31 compounds were found to possess good interaction at the active site. Among them, four squalene derivatives DQ-9, DQ-10, DQ-11, DQ-12, three biphenyl neolignans DQ-13, DQ-14, DQ-15and DS-17 showed the better affinity, which provide important ideas for finding novel leading compounds against cancer.Besides, nuclear receptor coactivator 2 (3up3) showed the significant activity compared with other targets,which indicates it may be the most effective target protein that would help to develop therapeutic drug against liver cancer. In addition, further cell level assaysor in-vivo experimental research for the confirmation of the compounds activity and bio-compatibility await further study in the future.
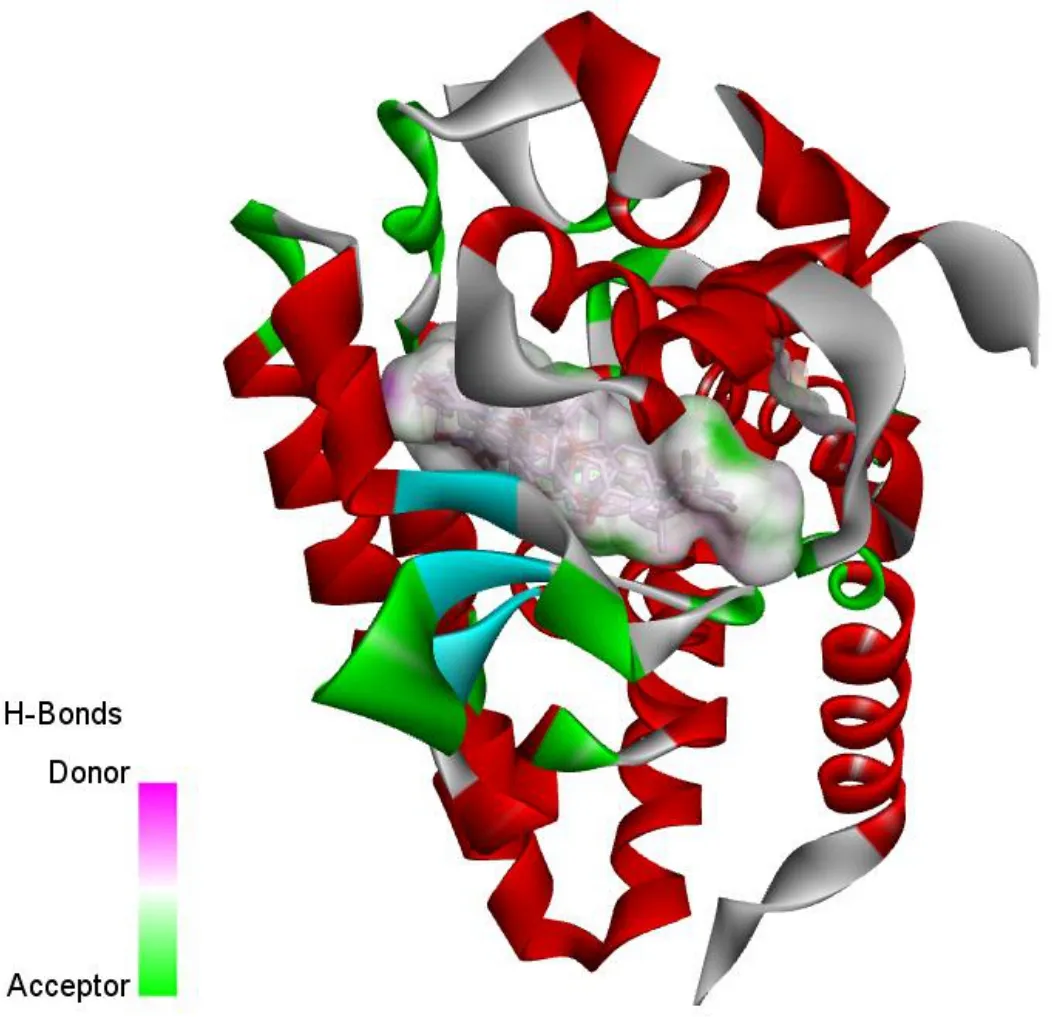
Fig. 3 The binding modes of the liver target structure (3up3) with the active compoents

Fig. 4 Close-up view of the active site residues of 3up3 bound with three potential compounds DQ-9, DQ-13, DS-17,respectively
[1] Okuda K, Ohtsuki T, Obata H, et al. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer, 1985, 56: 918-928.
[2] Newman D, Cragg G. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod,2012, 75: 311-335.
[3] Kuo PC, Shi LS, Damu AG, et al. Cytotoxic and antimalarial β-carboline alkaloids from the roots of Eurycoma longifolia. J Nat Prod, 2003, 66: 1324-1327.
[4] Park SJ, Nhiem NX, Kiem PV, et al. Five new quassinoids and cytotoxic constituents from the roots of Eurycoma longifolia. Bioorg Med Chem Lett, 2014, 24: 3835-3840.
[5] Hajjouli S, Chateauvieux S, Teiten MH, et al. Eurycomanone and eurycomanol from Eurycoma longifolia Jack as regulators of signaling pathways involved in proliferation,cell death and inflammation. Molecules, 2014, 19: 14649-14666.
[6] Chee FM, Rathinam X, Danial M, et al. Effects of methyljasmonate on 9-methoxycanthin-6-one content in Eurycoma longifolia (tongkat ali) root culture. Pak J Bot,2015, 47: 897-904.
[7] Chan KL, Choo CY, Abdullah NR, et al. Ethnopharmacol,2004, 92: 223-227.
[8] Chan KL, O'Neill MJ, Phillipson JD, et al. Plants as sources of antimalarial drugs. Part 3. Eurycoma longifolia. Planta Med, 1986, 52: 105-107.
[9] Fiaschetti G, Grotzer MA, Shalaby T, et al. Quassinoids:from traditional drugs to new cancer therapeutics. Curr Med Chem, 2011, 18: 316-328.
[10] Jiwajinda S, Santisopasri V, Murakami A, et al. In vitro anti-tumor promoting and anti-parasitic activities of the quassinoids from Eurycoma longifolia, a medicinal plant in Southeast Asia. Ethnopharmacol, 2002, 82: 55-58.
[11] Kuo PC, Damu AG, Lee KH, et al. Cytotoxic and antimalarial constituents from the roots of Eurycoma longifolia. Bioorg Med Chem, 2004, 12: 537-544.
[12] Razak MFA, Aidoo KE, Candlish AG. Mutagenic and cytotoxic properties of three herbal plants from Southeast Asia. Trop Biomed, 2007, 24: 49-59.
[13] Li ZC, Zhong WQ, Liu ZQ, et al. Large-scale identification of potential drugtargets based on thetopological features of human protein-protein interaction network. Analytica Chimica Acta, 2015, 871: 18-27.
[14] Boezio B, Audouze K, Ducrot P, et al. Network-based approaches in pharmacology. Molecular Informatics, 2017.
[15] Zhou WX, Cheng XR, Zhang YX. Institute of pharmacology and toxicology. 2012, 26: 4-9.
[16] Li ZC, Huang MH, Zhong WQ, et al. Identification of drug-target interaction from interactome network with"guilt-by-association" principle and topology features.Bioinformatics, 2015, 32: 1057-1064.
[17] Wang M, Zou H, Chen Q, et al. Isolation of new polyacetylenes from the roots of Eurycoma longifolia via high-speed counter-current chromatography. J Chromatogr B, 2017, s1055-1056: 39-44.
[18] Morita H, Kishi E, Takeya K, et al. Squalene derivatives from Eurycoma longifolia. Phytochemistry, 1993, 34: 765-771.
[19] Morita H, Kishi E, Takeya K, et al. Biphenylneolignans from wood of Eurycoma longifolia. Phytochemistry, 1992,31: 3993-3995.
[20] Takeda K, Hara N, Nishiyama T, et al. Corepressive function of nuclear receptor coactivator 2 in androgen receptor of prostate cancer cells treated with antiandrogen.Bmc Cancer, 2016, 16: 332.
[21] Ramanathan K, Veerakumar S, Amanulla SSD. Screening of potential plant compounds as survivin inhibitors and its anti-cancer efficacy by molecular docking. Current Enzyme Inhibition, 2017, 13: 1-8.
