基于UPLC最佳色谱参数的连翘中7个活性成分一测多评分析方法的建立及应用
2018-06-29刘章友周永峰张海珠
刘章友,李 杨,周永峰,张海珠*
(1.大理大学药学与化学学院,云南大理 671000;2.解放军第302医院全军中药研究所,北京 100039)
连翘,又名黄花条、黄奇丹等,是我国临床常用的传统中药之一,具有清热解毒、消痈散结、疏散风热等功效,被誉为“疮家圣药”〔1〕。2015年版《中华人民共和国药典》一部收载的正品连翘的来源为木犀科植物连翘Forsythia suspensa(Thunb.)Vahl的干燥果实,其主要分布在我国山西、河南、陕西、湖南等地。连翘的主要化学成分有连翘苷、连翘酯苷A、金丝桃苷、咖啡酸等,能够抑制肺炎双球菌、白喉杆菌、金黄色葡萄球菌〔2-4〕,抗菌作用显著;此外连翘还具有抗炎〔5-7〕、抗氧化〔8-10〕、保肝〔11〕等药理作用,临床常用于治疗急性风热感冒、痈肿疮毒等症。
目前,连翘药材的质量评价与控制主要是通过外标法测定连翘苷和连翘酯苷A的含量来实现〔12〕,但是常规外标法需要许多高纯度(>98.00%)的连翘苷和连翘酯苷A对照品作为对照,而这些高纯度对照品通常很难获得或者价格昂贵,因此限制了连翘药材的质量评控,且增加了检测成本。为解决这一困境,王智民等研发出一测多评分析方法(quantitative analysis of multi-component by a sin⁃gle-marker,QAMS)来解决这一问题,获得了越来越多的关注〔13-14〕。目前已有许多关于一测多评分析方法应用到中药材质量评控的报道,例如丹参、大黄〔15-16〕。虽然已有文献关于连翘中一测多评分析方法报道〔17〕,但是其仅仅测定了连翘的4个化学成分,并没有将连翘中含量极高的金丝桃苷和抑菌作用显著的绿原酸、咖啡酸、牛蒡子苷元等重要活性成分纳入测定范围;更重要的是,现有文献分析时间极长(超过110 min),未能达到满意的色谱分离度。
为了解决连翘中活性成分的含量测定过程中化学对照品缺乏和常规HPLC色谱分离度差的问题,本实验尝试建立基于超高效液相色谱(ultra-performance liquid chromatography,UPLC)的连翘中7个活性成分一测多评分析方法,以期为更加全面评价与控制连翘药材质量提供一个快速分析方法。
1 材料
1.1 仪器 Waters Acquity超高效液相色谱仪(Waters,USA);自动进样器(707,沃特世公司);光电二极管阵列检测器(PDA,沃特世科技上海有限公司);Empower 2色谱工作站(杭州瑞析科技有限公司);XS-205电子天平(Mettler toledo);AL-204电子天平(Mettler toledo);10~100 μL移液枪(上海艾研生物科技有限公司);超声仪(南京新辰生物科技有限公司,40 KHz);Milli-Q超纯水制备系统(Millipore,USA)。
1.2 试药与试剂 绿原酸对照品(批号151217,供含量测定用以99.68%计),咖啡酸对照品(批号151103,供含量测定用以99.40%计),芦丁对照品(批号151023,供含量测定用以98.94%计),金丝桃苷对照品(批号16081504,供含量测定用以98.34%计),连翘酯苷A对照品(批号151224,供含量测定用以98.31%计),连翘苷对照品(批号15102801,供含量测定用以98.54%计),牛蒡子苷元对照品(批号151029,供含量测定用以99.49%计),均购自成都吉欧特生物科技有限公司;色谱级甲醇(Thermo Fisher Scientific,USA);色谱级磷酸(85%w∕w,Thermo Fisher Scientific,USA)。
40批次连翘药材采集于山西、河南和河北等地,均为老翘。
2 方法与结果
2.1 色谱条件与系统适用性试验 色谱柱为ACQUITY UPLC HSS T3(2.1 mm×100 mm,1.8 μm),柱温为30℃。以乙腈(A)-0.1%磷酸水溶液(B)为流动相,梯度洗脱程序为:0~5 min:5%→8%(A);5~10 min:8%→10%(A);10~15 min:10%→15%(A);15~20 min:15%→30%(A);20~25 min:30%→50%(A);25~30min:50%→60%(A)。流速为0.3mL∕min;检测波长为275 nm;理论板数按连翘苷峰计算应不低于10 000。
2.2 对照品溶液的制备 精密称取绿原酸、咖啡酸、芦丁、金丝桃苷、连翘酯苷A、连翘苷和牛蒡子苷元对照品适量,精密称定,置于25 mL棕色容量瓶中,加色谱级甲醇制成每 1 mL含98、90、80、920、207、193、70 μg的溶液,即得。
2.3 供试品溶液的制备 取连翘药材,粉碎成细粉,准确称取0.20 g,置于25 mL容量瓶中,精密加入70%甲醇20 mL,密塞,称定重量,超声处理40 min(功率250 W,频率40 kHz),放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,用0.22 μm微孔滤膜滤过,取续滤液,即得。
2.4 色谱条件的优化 考虑到连翘药材70%甲醇溶液提取物中的化学成分极其复杂,针对目标成分想要获得满意的色谱分离度较为困难,尤其是针对芦丁、金丝桃苷、连翘苷、连翘酯苷A等4个活性成分,想要达到良好的色谱分离难以实现,因此在实验过程中通过考察流动相的流速和柱温的变化对目标成分的色谱分离参数(保留时间、分离度、对称因子、理论塔板数)的影响来确定最佳色谱条件。结果显示,当流动相的流速为0.3 mL∕min,柱温30℃的时候,供试品溶液中芦丁、金丝桃苷、连翘苷、连翘酯苷A等4个目标成分能够获得较为满意的色谱分离,PAD检测器检测结果显示,经过色谱参数优化后的4个目标成分色谱峰纯度达到阈值要求,表明获得了良好的色谱分离,保证了含量测定的准确性。经过优化后得到的典型的对照品溶液和供试品溶液色谱图见图1。
2.5 精密度考察 精密吸取混合对照品溶液2.0 μL连续进样6次,测定各个成分的色谱峰面积,计算RSD值。结果显示,绿原酸、咖啡酸、芦丁、金丝桃苷、连翘酯苷A、连翘苷和牛蒡子苷元色谱峰面积的RSD值分别为3.78%、4.61%、4.04%、3.91%、2.37%、1.99%、4.58%,表明仪器的精密度良好。
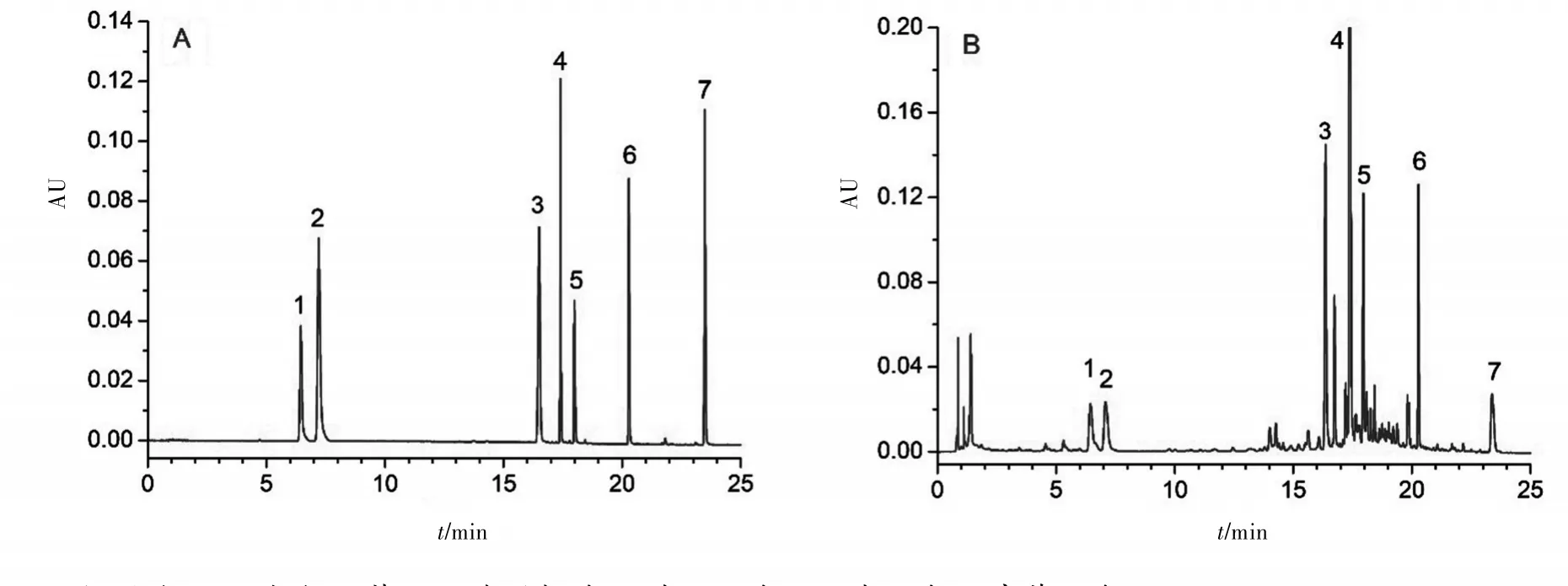
图1 典型的UPLC-UV图:A.混合对照品溶液;B.连翘供试品溶液
2.6 线性关系考察 分别精密吸取混合对照品溶液0.1、0.2、0.4、1.0、2.0、3.0 μL注入超高效液相色谱仪,按“2.1”项下色谱条件测定峰面积,以对照品进样量(x)为横坐标、峰面积(y)为纵坐标,得回归方程:y=5.0×106x+19 817(r=0.998 4);y=1.0×107x+14 085(r=0.998 9);y=4.6×105x+273 73(r=0.999 4);y=7.0× 106x+21 785(r=0.999 2);y=2.0× 106x+9 846(r=0.9995);y=3.0×106x+16 515(r=0.999 1);y=1.0×107x+24 832(r=0.999 2)。结果表明,绿原酸在0.009 80~0.196 00 μg范围内、咖啡酸在0.008 96~0.179 20 μg范围内、芦丁在0.00800~0.24000μg范围内、金丝桃苷在0.091 60~1.832 00 μg范围内、连翘酯苷A在0.02072~0.414 40 μg范围内、连翘苷在0.019 82~0.39640μg范围内、牛蒡子苷在0.00696~0.13920μg范围内线性关系良好。
2.7 稳定性考察 分别精密吸取混合对照品溶液和供试品溶液(供试品编号:yp01)在配制后的0、2、4、8、16、24 h测定峰面积,计算RSD值。结果显示混合对照品溶液中各个峰的峰面积RSD值分别为4.59%、3.86%、4.17%、3.79%、3.82%、4.09%、4.35%,供试品溶液中各个峰的峰面积RSD值分别为4.36%、2.48%、3.89%、4.92%、4.07%、3.29%、4.11%,表明对照品溶液和供试品溶液在制备后24 h内基本稳定,能够满足定量测定的要求。
2.8 重复性考察 取同一批次连翘药材细粉(供试品编号:yp01)约0.20 g,共6份,分别按“2.3”项下制备供试品溶液,按“2.1”项下色谱条件测定各个成分含量,结果显示绿原酸、咖啡酸、芦丁、金丝桃苷、连翘酯苷A、连翘苷和牛蒡子苷元的含量平均值分别为 0.23、0.06、1.29、63.52、19.10、12.31、0.14 mg∕g,RSD值分别为2.19%、3.08%、2.74%、3.19%、3.13%、2.59%、2.42%,表明方法的重现性良好。
2.9 加样回收率试验 精密称取连翘药材细粉(供试品编号:yp01)约0.10 g,共6份,置于25 mL棕色容量瓶中,精密加入绿原酸对照品溶液0.24 mL(浓度为84 μg∕mL)、咖啡酸对照品溶液0.13 mL(浓度为75 μg∕mL)、芦丁对照品溶液0.87 mL(浓度为150 μg∕mL)、金丝桃苷对照品溶液6.68 mL(浓度为950 μg∕mL)、连翘酯苷A对照品溶液2.43 mL(浓度为785 μg∕mL)、连翘苷对照品溶液 1.47 mL(浓度为835 μg∕mL)、牛蒡子苷元对照品溶液0.50 mL(浓度为20 μg∕mL),最后精密加入70%甲醇溶液7.68 mL,分别按“2.3”项下方法制备供试品溶液、“2.1”项下色谱条件测定各个成分含量,按公式(1)计算回收率,结果显示7个成分的回收率平均值为95.4%、96.3%、97.2%、102.6%、103.7%、98.5%、95.1%,表明方法的准确性较好。

2.10 内标物质的选择、校正因子的测定和校正因子的耐用性考察 芦丁是连翘药材中一种价廉易得、且药理作用广泛的活性成分。同时在前期预实验中发现芦丁的保留时间处于7个分析物的中间时段,保留时间适中,因此在实验中,尝试选用芦丁作为内标物质。以芦丁为内标,根据公式(2)计算校正因子,见表1。同时为了考察计算得到的校正因子的稳健性,实验中通过改变柱温和流速来评价校正因子的稳健性,结果见表2。

其中As代表内标物的色谱峰面积,Cs代表内标物质的进样浓度,Ai代表成分i的色谱峰面积,Ci代表样品溶液中成分i的浓度。通过公式(2)我们能够推导出公式(3):

通过公式(3)可以计算其他被测化合物的浓度。
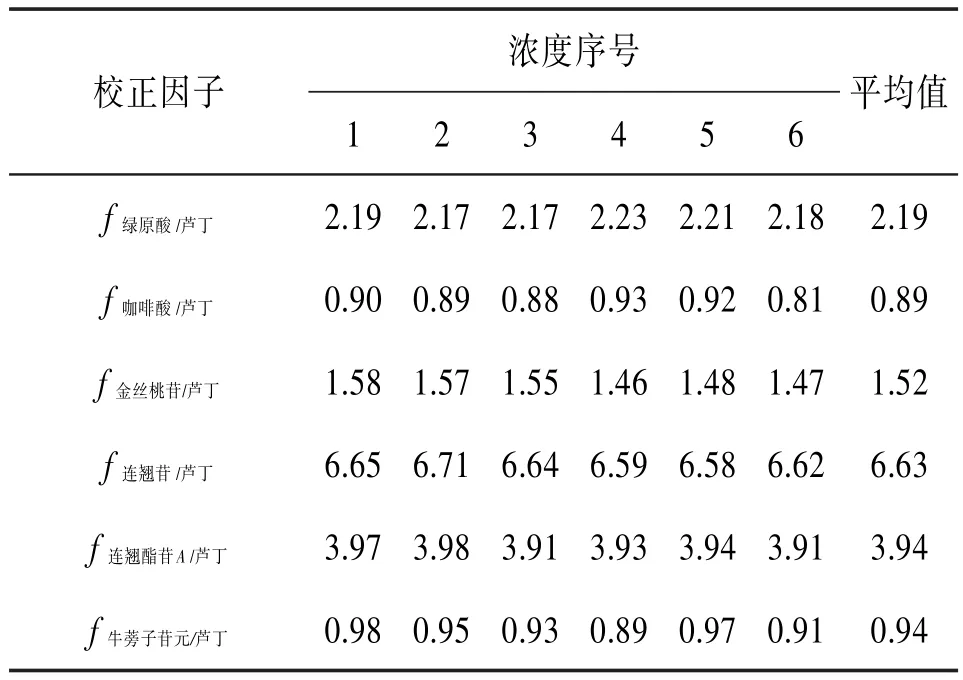
表1 相对校正因子测定结果
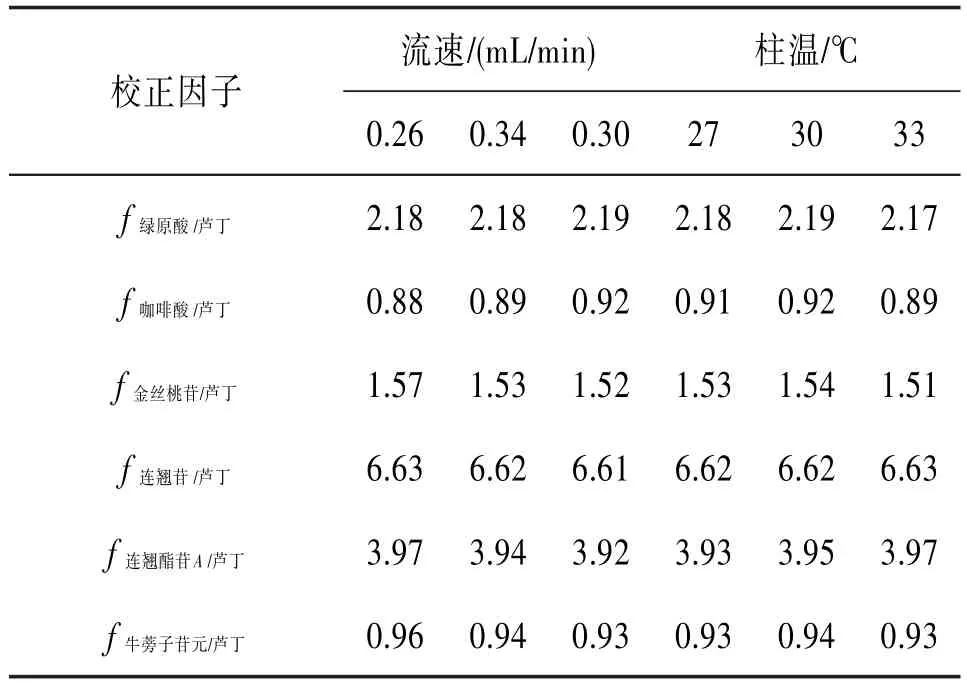
表2 影响因子的稳健性考察结果(流速和柱温对校正因子的影响)
2.11 供试品的分析及常规外标法和一测多评法的准确性比较 40批次连翘药材按照供试品溶液制备方法制备供试品溶液,精密吸取混合对照品溶液与供试品溶液各2.0 μL注入液相色谱仪测定,记录色谱峰面积,按照外标法和新建立的一测多评方法对绿原酸等7个化学成分进行含量测定,部分供试品测定结果见表3。结果显示两种方法测定的结果相对偏差较小,除了绿原酸和咖啡酸的准确性稍微差一点外(在275 nm处紫外吸收较小),其余5个成分通过一测多评分析方法测定的准确性较高。

表3 一测多评法和外标法测定结果对比(mg∕g,n=3)

续表3
采用Heml-1.0软件进行热图(heat-map anal⁃ysis)和聚类分析(cluster analysis),采用统计软件SIMCA-P 13.0进行主成分分析(principal component analysis,PCA)。聚类分析和PCA分析可以初步区分开不同产地连翘药材样品,大致可以得到3类:得分图清晰地显示yp01~yp18为一类(样品产地为山西),yp19~29为一类(样品产地为河南,除了yp30处于离群外),yp31~yp40为一类(样品产地为河北)。分析结果表明不同产地的连翘药材从化学成分差异的角度来看确实存在一定的品质差异,而这种差异主要由其连翘苷、连翘酯苷A、金丝桃苷、芦丁等化学成分的含量所体现的。这也表明尽可能多地检测连翘中的活性成分对于评价不同来源的连翘药材质量差异具有重要的现实意义。聚类分析图和主成分分析图见图2。
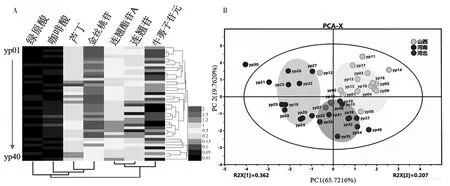
图2 统计学分析结果:A.以40批次连翘样品中7个化学成分含量的聚类分析和热图分析;B.以40个批次连翘样品中7个化学成分含量的主成分分析
3 讨论
本实验通过优化UPLC色谱参数建立了检测连翘中7个活性成分含量的一测多评分析方法并进行了方法学评价。该方法相对于传统质量控制方法,能够对多种成分进行同步质量控制,同时又能克服对照品紧缺和检测成本高的困难〔14〕。
实验过程中对甲醇-0.1%磷酸水、乙腈-0.1%磷酸水流动相体系进行了比较,结果显示采用乙腈-0.1%磷酸水的流动相体系,供试品溶液中目标峰与杂质峰的分离度较好,同时基线平稳,因此选用乙腈-0.1%磷酸水作为流动相。采用PAD检测器对7个标准品溶液在210~420 nm范围内进行全波长扫描,结果显示7个化学成分在277 nm和235 nm波长处均具有较强吸收,但在235 nm波长下,供试品溶液色谱图中杂质峰较多,对目标峰干扰严重,色谱分离度难以达到要求,最后优选以277 nm作为检测波长。
对芦丁、金丝桃苷、连翘酯A、连翘苷的色谱分离参数进行优化过程中,随着柱温从20℃增加到35℃,目标成分的保留时间和对称因子逐渐减小,分离度和理论塔板数逐渐增大,综合考虑4个目标成分的色谱分离参数变化趋势、系统的压力等因素,最后优选30℃作为最佳柱温。随着流动相流速从0.1 mL∕min增加到0.4 mL∕min过程中,目标成分的保留时间、对称因子逐渐减小,分离度和理论塔板数逐渐增大,但在0.3 mL∕min时,金丝桃苷、连翘酯A和连翘苷的理论塔板数有减小的趋势,同时当流速增加到0.3 mL∕min时,系统的压力已经超过12 000 psi,综合考虑4个目标成分的色谱分离参数变化趋势、系统的压力等因素,最后优选0.3 mL∕min作为最佳流速。
在实验过程中,由于采用的是Waters Acquity UPLC和HSS T3色谱柱,缺少不同厂家的同类型色谱仪器,暂时无法考察测得的校正因子用于不同厂家不同型号色谱仪器和色谱柱的耐用性,因此考察了不同流速和色谱柱温对校正因子的影响,结果显示,测得的校正因子具有较好的稳健性。
在实验中,对QAMS分别进行精密度考察、线性关系考察、稳定性考察、重复性考察、加样回收率试验等方法学考察。结果显示,连翘中的7种活性成分线性关系良好,且重现性好,能满足测量要求。
综上所述,本研究建立了可靠的测定连翘中7个活性成分含量的分析方法,为后续连翘获悉成分含量的研究提供了基础。
〔1〕张廷模.临床中药学〔M〕.上海:上海科学技术出版社,2006:109.
〔2〕段文娟,耿岩玲,祝贺,等.中药连翘化学成分和分析方法的研究进展〔J〕.山东科学,2010,23(2):33-37.
〔3〕宋小俊.连翘不同部位化学成分研究进展〔J〕.西北药学杂志,2014,29(2):220-222.
〔4〕胡静,马琳,张坚,等.连翘的研究进展〔J〕.中南药学,2012,10(10):760-764.
〔5〕KANG H S,LEE J Y,KIM C J.Anti-inflammatory activity of arctigenin from Forsythiae Fructus〔J〕.J Ethnopharma⁃col,2008,116(2):305-312.
〔6〕MULUYE R A,BIAN Y,ALEMU P N.Anti-inflammatory and Antimicrobial Effects of Heat-clearing Chinese Herbs:A Current Review〔J〕.J Tradit Complement Med,2014,4(2):93-98.
〔7〕芮菁,尾崎幸纮,唐元泰.连翘提取物的抗炎镇痛作用〔J〕.中草药,1999,30(1):43-45.
〔8〕耿慧君,王文科,毕润成.连翘叶的体外抗氧化活性研究〔J〕.山西师范大学学报(自然科学版),2005,19(4):71-73.
〔9〕LU T,PIAO X L,ZHANG Q,et al.Protective effects of For⁃sythia suspensa extract against oxidative stress induced by diquat in rats〔J〕.Food Chem Toxicol,2010,48(2):764-770.
〔10〕CHANG M J,HUNG T M,MIN B S,et al.Lignans from the Fruits of Forsythia suspensa(Thunb.) Vahl Protect High-Density Lipoprotein during Oxidative Stress〔J〕.Biosci Biotechnol Biochem,2008,72(10):2750-2755.
〔11〕徐春媚,王文生,曹艳红,等.连翘护肝作用的实验研究〔J〕.黑龙江医药科学,2001,24(1):10.
〔12〕国家药典委员会.中华人民共和国药典:一部〔M〕.北京:中国医药科技出版社,2015:171.
〔13〕王智民,高慧敏,付雪涛,等.“一测多评”法中药质量评价模式方法学研究〔J〕.中国中药杂志,2006,31(23):1925-1928.
〔14〕王智民,钱忠直,张启伟,等.一测多评法建立的技术指南〔J〕.中国中药杂志,2011,36(6):657-658.
〔15〕杨菲,王智民,张启伟,等.“一测多评”法测定丹参酚酸类成分的含量〔J〕.中国中药杂志,2011,36(17):2372-2379.
〔16〕TAN P,ZHANG L,ZHAO Y L,et al.A practical method for the simultaneous quantitative determination of twelve anthraquinone derivatives in rhubarb by a single-marker based on ultra-performance liquid chromatography and chemometric analysis〔J〕.Analtical Methods,2016,8(19):3927-3934.
〔17〕孔晶晶,朱晶晶,王智民,等.一测多评法测定连翘中多种不同类型成分的含量〔J〕.中国药学杂志,2010,45(17):1301-1304.
