下调MLAA-22基因对U937细胞增殖与分化的影响
2018-06-25张王刚
崔 鹤,张王刚
(1. 内蒙古医科大学附属医院血液内科,内蒙古呼和浩特 010059;2.西安交通大学第二附属医院血液内科,陕西西安 710004)
急性单核细胞白血病是急性髓细胞白血病(acute myeloid leukemia, AML)中的一个亚型,按照法、美、英(France-America-Britain, FAB)协作组分型标准,该亚型被称为M5型。在西方国家,M5型约占AML的8%;在我国,M5型占AML的20%~30%,仅次于M2型[1]。M5型患者可经常出现高白细胞血症、髓外浸润、弥漫性血管内凝血等临床表现[2],白血病细胞表面常有CD11b及CD56表达[1],还可伴有t(9;11) (p22;q24)、t(9;11) (p22;q23)、t(9;11) (q33;q23)、t(11;17) (q23;q21)、t(11;17) (q23;q25)、t(11;17) (q23;q12)、t(11;17) (q24;q21)、t(11;17) (123;q11-21)、t(11;20) (p15;q11.2)、t(11;20) (p15;q11)、t(1;11) (p31;q23)、t(1;11;4) (q21;q23;p16)、t(8;16) (p11;p13)、t(8;22) (p11;q13)等多种染色体核型异常[3-15]。随着联合化疗方案的优化、支持治疗方案和造血干细胞移植方法的改进,使M5型患者的疗效较前有所提高,但如何有效清除微小残留病变以预防复发,以及如何改善复发/难治性患者的疗效仍是目前难以解决的两大问题。
目前,越来越多的生物治疗方法应用到肿瘤的治疗当中,该方法已成为唯一有希望完全消灭肿瘤细胞的治疗方法,其中免疫治疗方法倍受人们关注。为了研究急性单核细胞白血病的免疫治疗方法,我们实验组在2005年采用重组cDNA表达文库血清学分析法筛选出了一系列急性单核细胞白血病相关抗原(monocytic leukemia-associated antigen, MLAA)基因,MLAA-22基因是其中之一[16]。我们前期研究发现,该基因是一个抗凋亡基因[17-19],在初诊急性单核细胞白血病患者中的表达高于正常人[20]。为了进一步明确该基因的生物学功能,我们采用规律成簇间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)/CRISPR相关核酸酶9(CRISPR-associated 9, Cas9)系统下调U937细胞中MLAA-22基因的表达,初步研究了该基因下调后对U937细胞增殖和分化的影响。
1 材料与方法
1.1主要试剂与细胞系胎牛血清购自美国Gibco公司;RPMI-1640培养基购自美国Hyclone公司;Trizol试剂购自美国Invitrogen公司;反转录试剂盒和化学发光检测试剂盒购自美国Thermo Fisher公司;实时荧光定量PCR试剂盒购自瑞士Roche公司;感染增强液购自上海吉凯基因化学技术有限公司;十二烷基硫酸钠—聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis, SDS-PAGE)凝胶配制试剂盒购自上海碧云天生物技术有限公司;琼脂糖凝胶DNA纯化回收试剂盒购自天根生化科技有限公司;基因敲除和突变检测试剂盒购自上海吉盛医学科技有限公司;感染增强液、lenti-Cas9-puro、lenti-sgRNA-EGFP及阴性对照病毒购自上海吉凯基因化学技术有限公司,pMD19-T载体克隆试剂盒、大肠杆菌TOP10、5-溴-4-氯-3-吲哚-D-半乳糖苷(X-Gal)、异丙基-β-D-硫代吡喃半乳糖苷(IPTG)购自日本TaKaRa公司;U937细胞由西安交通大学第二附属医院血液内科保存;其余试剂为国产分析纯。
1.2细胞培养使用含100 mL/L胎牛血清的RPMI-1640培养基培养U937细胞,培养条件为37 ℃、饱和湿度、50 mL/L的CO2。
1.3采用CRISPR/Cas9系统下调U937细胞MLAA-22基因CRISPR/Cas9系统由2种慢病毒组成(lenti-Cas9-puro及lenti-sgRNA-EGFP),它们分别向U937细胞中导入核酸内切酶Cas9基因和sgRNA,从而实现对目的基因的下调。其中lenti-cas9-puro携带嘌呤霉素标记,lenti-sgRNA-EGFP携带绿色荧光标记,其中sgRNA的序列为:5′-GGAGTTTGCTGGAAATCCAC-3′。
1.3.1Lenti-Cas9-puro-U937细胞株的建立 收集处于对数生长期的U937细胞,1 000 r/min,离心5 min,使用感染增强液重悬细胞,制成细胞密度为5×104个/mL的细胞悬液,接种到48孔培养板中,每孔200 μL,在每孔加入一定体积的聚凝胺(终浓度5 μg/mL),加入适量Lenti-Cas9-puro感染U937细胞(感染复数:20);感染后16 h,将各孔中细胞收集到1.5 mL EP管中,2 000 r/min,离心2 min,去掉上清液,更换为500 μL完全培养基,轻轻混匀后在24孔板中继续培养;感染后72 h,每孔加入终浓度为4 μg/mL的嘌呤霉素,继续培养48 h;将各孔中细胞收集到1.5 mL EP管中,2 000 r/min,离心2 min,去掉上清液,更换为500 μL完全培养基,每孔加入终浓度为2 μg/mL的嘌呤霉素继续培养,待细胞状态良好时进行下游实验,同时冻存一部分细胞用于后续实验。
1.3.2采用Lenti-sgRNA-EGFP感染Lenti-Cas9-puro-U937细胞 实验分组情况:MLAA基因下调组(即感染Lenti-sgRNA-EGFP后的Lenti-Cas9-puro-U937细胞,简称KD组)、阴性对照组(即感染阴性对照病毒的U937细胞,简称NC组)、空白对照组(即U937细胞,简称CON组)。收集处于对数生长期的Lenti-Cas9-puro-U937细胞,1 000 r/min,离心5 min,使用感染增强剂重悬细胞(细胞密度5×104个/mL),接种到48孔培养板中,每孔200 μL,在每孔加入一定体积的聚凝胺(终浓度5 μg/mL),加入适量Lenti-sgRNA-EGFP(感染复数:20);感染后16 h,将各孔中细胞收集到1.5 mL EP管中,2 000 r/min,离心2 min,去掉上清液,更换为500 μL完全培养基,轻轻混匀后在24孔板中继续培养;感染后96 h,使用荧光显微镜观察绿色荧光蛋白的表达情况,计算感染效率。
1.3.3CruiserTM酶切法检测sgRNA的活性 在感染Lenti-sgRNA-EGFP后的120 h收集细胞,按照基因组DNA提取试剂盒操作说明抽提样品的基因组DNA,以抽提的基因组DNA为模板对MLAA-22基因突变区进行PCR扩增(反应条件:预变性,94 ℃ 90 s;变性,94 ℃ 30 s;退火,60 ℃ 30 s;延伸,72 ℃ 30 s;共30个循环,终止延伸,72 ℃ 30 s)。PCR扩增所需引物为:5′-TTCAGGAACGACCTTTTGCAAGG-3′(上游);5′-GGCAGACAGGTCAGAAACTTT-3′(下游)。将获得的PCR产物进行琼脂糖凝胶电泳,接着按照琼脂糖凝胶DNA纯化回收试剂盒的操作说明回收及纯化PCR产物,随后按照基因敲除和突变检测试剂盒操作说明配制CruiserTM酶切反应体系,45 ℃ 反应20 min后,立即加入2 μL终止液,最后将酶切产物进行琼脂糖凝胶电泳。
1.3.4基因突变区PCR产物的TA克隆及测序 在无菌PCR管中加入TA克隆反应体系(pMD19-T载体:1.0 μL,PCR产物1.0 μL,双蒸水3.0 μL);在TA克隆反应体系中加入5 μL溶液1;16 ℃反应30 min;将产物加入到大肠杆菌TOP10感受态细胞(100 μL)中,放置于冰上30 min;在42 ℃金属浴中放置90 s,然后放置于冰上2 min;加入含氨苄青霉素(终浓度100 μg/mL)的LB液体培养基(500 μL)中,置于37 ℃摇床振荡培养1 h;取适量菌液均匀涂布在含X-Gal(终浓度0.4 mmol/L)、IPTG(终浓度0.5 mmol/L)、氨苄青霉素(终浓度100 μg/mL)的LB固体培养基平板上,在恒温培养箱中倒置培养16 h,随机挑取31个白色单克隆菌落送生工生物工程(上海)股份有限公司进行测序,运用BLAST软件对测序结果进行分析,计算MLAA-22基因的突变率。
1.4下调MLAA-22基因对U937细胞增殖的影响使用Lenti-sgRNA-EGFP感染Lenti-Cas9-puro-U937细胞,96 h后收集待测细胞,使用完全培养基重悬细胞,并进行细胞计数;将细胞均匀铺于96孔板中(细胞密度2×103个/孔),每孔100 μL,每组均设立3个重复孔;铺好细胞后,待细胞完全沉淀下来,放入37 ℃、50 mL/L CO2细胞培养箱中培养;继续培养24、48、72、96、120 h时终止培养,在培养终止前4 h时加入10 μL CCK-8于待测孔中;将待测细胞重新放入37 ℃、饱和湿度、50 mL/L CO2细胞培养箱继续培养,4 h后将96孔板置于振荡器上振荡5 min,使用酶标仪在450 nm波长检测待测样品的吸光度值。以上实验重复3次。
1.5下调MLAA-22基因对U937细胞分化的影响使用Lenti-sgRNA-EGFP感染Lenti-Cas9-puro-U937细胞,感染120 h后收集待测细胞,1 000 r/min、离心3 min,弃去上清;PBS洗涤细胞沉淀1次,1 000 r/min,离心3 min,弃去上清;使用1×结合缓冲液洗涤细胞沉淀1次,1 000 r/min,离心3 min,弃去上清;加入PBS重悬细胞沉淀(细胞密度5×105个/mL);加入20 μL抗CD11b抗体,室温、避光孵育30 min;1 000 r/min,离心3 min,收集细胞,使用1 mL PBS洗涤细胞沉淀3次,1 000 r/min,离心3 min,收集细胞;使用800 μL 1×细胞染色缓冲液重悬细胞并混匀,转移至96孔板中,每孔200 μL,使用流式细胞仪检测CD11b阳性细胞率。
1.6统计学分析使用SPSS 18.0统计软件对数据进行分析,所有实验数值描述为均值±标准差,采用t检验进行样本之间的比较,当P<0.05时,认为两样本之间存在统计学差异。
2 结 果
2.1感染Lenti-sgRNA-EGFP细胞的绿色荧光蛋白表达率感染Lenti-sgRNA-EGFP后96 h,使用荧光显微镜下观察各组细胞绿色荧光蛋白的表达情况。结果显示,NC组和KD组的绿色荧光表达率均>90%(图1),说明Lenti-sgRNA-EGFP的感染效率高,可以继续进行下游实验。

图1 感染Lenti-sgRNA-EGFP后细胞的绿色荧光蛋白表达情况Fig.1 Expression of green fluorescent protein in the cells transfected with lentivirus-sgRNA-EGFP (×200)
2.2CruiserTM酶切法检测sgRNA的活性感染Lenti-sgRNA-EGFP后120 h,收集细胞,抽提样品的基因组DNA,基因组DNA经PCR扩增后,使用琼脂糖凝胶电泳检测PCR产物大小,结果如图2A所示:PCR产物大小为1 050 bp,与预测值一致;PCR产物经CruiserTM酶切,酶切产物的琼脂糖凝胶电泳结果如图2B所示:PCR产物被CruiserTM酶切割为大小分别为645 bp和405 bp的2条片段,条带大小与预测值一致,这说明本文中所采用的CRISPR/Cas9系统中的sgRNA序列能够高效、准确地识别基因编辑靶点。

图2 CruiserTM酶切检测sgRNA的活性Fig.2 Detection of sgRNA’s activity by CruiserTM enzyme digestion
2.3PCR产物的TA克隆及测序将经胶回收试剂盒回收、纯化后获得的KD组细胞的MLAA-22基因突变区的PCR产物与T载体连接,并将连接产物转化到大肠杆菌TOP10感受态细胞中,经蓝白斑筛选,随机送31个阳性单克隆菌落生工生物工程(上海)股份有限公司进行测序,采用BLAST软件对测序结果进行分析,分析结果(图3)显示:在31个单克隆菌落中,有19个单克隆菌落发生了基因突变,MLAA-22基因突变率为61.3%(其中第1~13号单克隆菌落发生了碱基缺失;第14号及第16号单克隆菌落同时发生了碱基置换和碱基缺失;第17~19号单克隆菌落同时发生了碱基置换和碱基插入)。说明本研究中所采用的CRISPR/Cas9系统可用于后续的实验研究。

图3 PCR产物的TA克隆及测序结果Fig.3 TA cloning and sequencing of PCR products
2.4下调MLAA-22基因对U937细胞增殖的影响为了研究MLAA-22基因的下调对U937细胞增殖的影响,本实验使用CCK-8试剂盒分别检测CON、NC及KD组的U937细胞的增殖情况,绘制生长曲线,比较各组细胞的增殖能力。结果显示:与CON和NC组相比,KD组细胞的增殖速度明显小于CON和NC组(P<0.01);CON和NC组细胞的增殖速度无明显差异(P>0.05,图4)。这说明MLAA-22基因的下调明显抑制了U937细胞的增殖。
2.5下调MLAA-22基因对U937细胞分化的影响下调U937细胞MLAA-22基因表达后,使用流式细胞仪检测细胞表面分化抗原CD11b的表达情况。结果如图5所示:KD组CD11b阳性细胞率高于NC组和CON组(P<0.01),NC组CD11b阳性细胞率和CON组无明显差别(P>0.05)。这说明,下调MLAA-22基因的表达可增加细胞表面分化抗原CD11b的表达,促进了U937细胞向成熟方向分化。

图4 下调MLAA-22基因对U937细胞增殖的影响Fig.4 Cell proliferation in MLAA-22-knockdown U937 cells
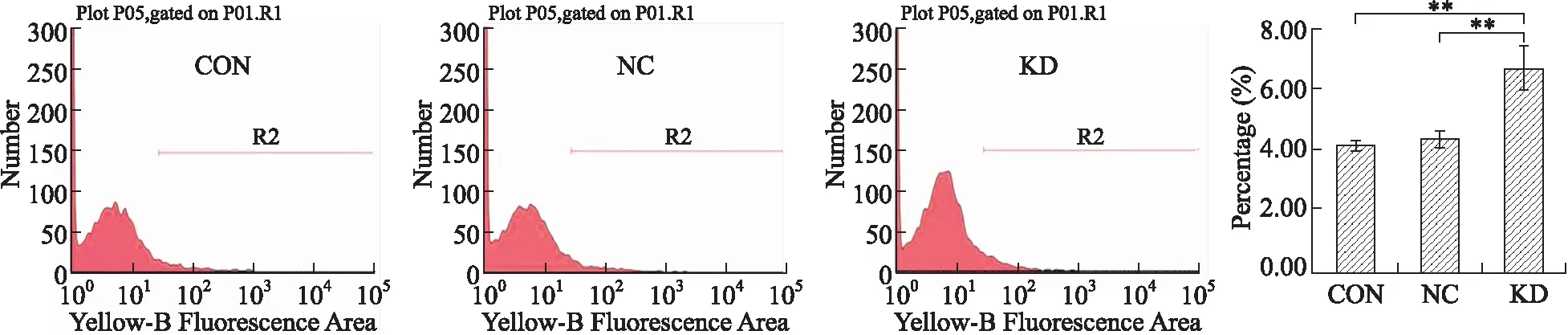
图5 下调MLAA-22基因对U937细胞分化的影响Fig.5 Cell differentiation in MLAA-22-knockdown U937 cells
3 讨 论
基因编辑技术是常用于基因功能和基因治疗研究的、对基因组进行定点修饰的技术[21]。CRISPR/Cas9系统是一种最新的、高效的基因编辑技术。它由1条能够与靶基因序列碱基互补配对结合的sgRNA序列和1种能够切割靶基因DNA双链的核酸内切酶(Cas9蛋白)组成[22],sgRNA负责识别靶基因的DNA序列。需要注意的是,被sgRNA识别的DNA序列的上游必须存在一PAM序列,通常为5′-NGG,它的作用是引导Cas9蛋白与靶基因结合[23]。靶基因的DNA序列被CRISPR/Cas9系统切断后,将会立即通过机体细胞内存在的非同源末端连接(non-homologous end joining, NHEJ)和同源定向修复(homology directed repair, HDR)2种修复途径进行自我修复[24]:NHEJ是不依赖于同源DNA序列的修复方式,修复的结果是在DNA切割位点上产生短的插入和缺失突变,能够引起靶基因的开放阅读框发生移位突变,是处于G0和G1期的细胞基因修复的主要途径;HDR是依赖于同源DNA序列的修复方式,可以对靶基因进行精确的编辑或引入特定的基因序列改变,是处于S和G2期的细胞基因修复的主要途径[25-26]。最终CRISPR/Cas9系统借助于这2种机体细胞内部固有的基因修复途径实现对目的基因重新编辑。
细胞增殖失控、凋亡受阻、分化障碍是白血病细胞的重要特征,其中细胞凋亡受阻和分化障碍是引起白血病细胞增殖失控的2个重要因素。本课题组前期研究发现MLAA-22基因具有抗凋亡的生物学功能。为了明确该基因是否还具有调控细胞分化的生物学功能,本研究应用CRISPR/Cas9系统在U937细胞中下调了MLAA-22基因的表达,TA克隆及测序结果显示,MLAA-22基因突变率为61.3%,这说明本文所使用的CRISPR/Cas9系统可有效引起MLAA-22基因突变,可用于后续的实验研究。此外,细胞增殖检测结果显示,MLAA-22基因的表达下调后,U937细胞增殖速度明显减慢;而且细胞分化检测结果显示,MLAA-22基因的表达下调后,U937细胞表面分化抗原CD11b阳性细胞率增加,促进了U937细胞向成熟方向分化。这说明MLAA-22基因可能通过调控U937细胞分化而引起细胞增殖失控,从而促进急性单核细胞白血病的发生与发展。本研究结果为将来深入研究该基因的生物学功能的分子作用机制以及靶向治疗药物奠定了基础。
参考文献:
[1] 胡昳歆. 急性单核细胞白血病预后因素研究进展[J]. 国际输血及血液学杂志, 2012, 35(1):62-65.
[2] POHLMANN R, BHARGAVA P. Acute monoblastic leukemia with abnormal granules and intravascular coagulation: Diagnostic pitfalls[J]. Am J Hematol, 2009, 84(11):773-775.
[3] DEWALD GW, MORRISON-DELAP SJ, SCHUCHARD KA, et al. A possible specific chromosome marker for Monocytic leukemia:three more patients with t(9;11)(p22;q24) and another with t(11;17)(q24;q21), each with acute monoblastic leukemia[J]. Cancer Genet Cytogenet, 1983, 8(3):203-212.
[4] DIAZ MO, LE BEAU MM, PITHA P, et al. Interferon and c-ets-1 genes in the translocation (9;11)(p22;q23) in human acute monocytic leukemia[J]. Science, 1986, 231(4735):265-267.
[5] LE CONIAT M, ROMANA SP, BERNARD O, et al. A novel translocation, t(9;11)(q33;q23) involving the HRX gene in an acute monocytic leukemia[J]. C R Acad Sci III, 1993, 316(7):692-697.
[6] KIM KE, KIM SH, HAN JY. Acute monocytic leukemia with t(11;17)(q23;q21) involving a rearrangement of mixed lineage leukemia gene[J]. Korean J Lab Med, 2006, 26(5):329-333.
[7] KUROSU T, TSUJI K, OHKI M, et al. A variant-type MLL/SEPT9 fusion transcript in adultde novo acute monocytic leukemia (M5b) with t(11;17)(q23;q25)[J]. Int J Hematol, 2008, 88:192-196.
[8] SHEKHTER-LEVIN S, GOLLIN SM, KAPLAN SS, et al. Involvement of the MLL and RAR alpha genes in a patient with acute monocytic leukemia with t(11;17)(q23;q12)[J]. Leukemia, 2000, 14(2):520-522.
[9] REEVES BR, KEMPSKI H, JANI K, et al. A case of acute monocytic leukemia with t(11;17) involving a rearrangement of mLL-1 and a region proximal to the RARA gene[J]. Cancer Genet Cytogenet, 1994, 74(1):50-53.
[10] KAKAZU N, SHINZATO I, ARAI Y, et al. Involvement of the NUP98 gene in a chromosomal translocation t(11;20)(p15;q11.2) in a patient with acute monocytic leukemia (FAB-M5b)[J]. Int J Hematol, 2001, 74(1):53-57.
[11] CHEN S, XUE Y, CHEN Z, et al. Generation of the NUP98-TOP1 fusion transcript by the t(11;20) (p15;q11) in a case of acute monocytic leukemia[J]. Cancer Genet Cytogenet, 2003, 140(2):153-156.
[12] NÖLLE I, SCHLEGELBERGER B, SCHMITZ N, et al. Acute monocytic leukemia with translocation t(1;11)(p31;q23): Simultaneous staining of chromosomes and cell surface antigens[J]. Haematol Blood Transfus, 1990, 33:145-149.
[13] SO CW, MA SK, WAN TS, et al. Analysis of MLL-derived transcripts in infant acute monocytic leukemia with a complex translocation (1;11;4)(q21;q23;p16)[J]. Cancer Genet Cytogenet, 2000, 117(1):24-27.
[14] HANADA T, ONO I, MINOSAKI Y, et al. Translocation t(8;16)(p11;p13) in neonatal acute monocytic leukaemia[J]. Eur J Pediatr, 1991, 150(5):323-324.
[15] KITABAYASHI I, AIKAWA Y, YOKOYAMA A, et al. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation[J]. Leukemia, 2001, 15(1):89-94.
[16] CHENG, ZHANG WG, CAO XM, et al. Serological identification of immunogenic antigens in acute monocytic leukemia[J]. Leuk Res, 2005, 29(5):503-509.
[17] CUI H, LAN X, LU SM, et al. Bioinformatic prediction and functional characterization of human KIAA0100 gene[J]. J Pharm Anal, 2017, 7(1):10-18.
[18] CUI H, LAN X, LU SM, et al. Preparation of monoclonal antibody against human KIAA0100 protein and northern blot analysis of human KIAA0100 gene[J]. J Pharm Anal, 2017, 7(3):190-195.
[19] 崔鹤,张王刚. 抗人KIAA0100蛋白多克隆抗体的制备[J]. 现代肿瘤医学, 2017, 25(5):681-686.
[20] ZHOU FL, ZHANG WG, MENG X, et al. Bioinformatic analysis and identification for a novel antigen MLAA-22 in acute monocytic leukemia[J]. J Exp Hematol Chin Assoc Pathophysiol, 2008, 16(3):466-471.
[21] 王丹,周美亮,李金博,等. CRISPR/Cas9基因组编辑技术及其在草类植物中的应用[J]. 草业科学, 2017, 34(6):1204-1214.
[22] MELODY R, ANDREW K, CAROLINE W, et al. What is CRISPR/Cas9?[J]. Arch Dis Child Educ Pract Ed, 2016, 101(4):213-215.
[23] HSU PD, SCOTT DA, WEINSTEIN JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases[J]. Nat Biotechnol, 2013, 31(9):827-832.
[24] SHEN S, LOH TJ, SHEN H, et al. CRISPR as a strong gene editing tool[J]. Bmb Reports, 2017, 50(1):20-24.
[25] KANNAN R, VENTURA A. The CRISPR revolution and its impact on cancer research[J]. Swiss Med Wkly, 2015, 145:w14230.
[26] SHUMAN S, GLICKMAN MS. Bacterial DNA repair by non-homologous end joining[J]. Nat Rev Microbiol, 2007, 5(11):852-861.
