纳米乳化油强化硝酸盐反硝化产气变化研究
2018-06-25连玉倩何江涛何宝南中国地质大学北京水资源与环境工程北京市重点实验室北京100083
连玉倩,何江涛,梁 雨,何宝南 (中国地质大学(北京),水资源与环境工程北京市重点实验室,北京 100083)
原位生物处理技术是处理地下水中污染物的一种廉价、高效率的方法[1],而投加碳源作为电子供体可以强化土壤和地下水中微生物去除污染物的能力[2-3].目前,因乳化油操作运行费用低,且是食品级物质不会对环境造成二次污染[4-6]国内外学者将其作为一种治理地下水污染物的新兴碳源.此外,乳化油是一种缓慢释放基质,具有溶解度低.在含水层中迁移性良好等优点[7-9].近年来乳化油用于原位去除地下水硝酸盐、trichloroethene (TCE)、高氯酸盐、重金属U(Ⅵ)、Cr(Ⅵ)等污染物取得了很好的治理效果[10-16],但是在实际应用过程中也发现了新的问题,其中主要问题之一即为:乳化油反应带强化修复地下水污染过程中,往往会造成含水层介质渗透性损失,严重时甚至会发生堵塞,这种情况一旦产生,受污染的地下水流经反应带时会产生绕流现象,从而导致修复效率降低或者失效.
已有研究表明,乳化油碳源原位生物修复地下水污染时造成含水层渗透性损失原因主要包括:乳化油在多孔介质中的吸附截留及其粒径大于含水层多孔介质孔径时产生的物理截留、微生物的大量增殖和生物量的积累造成的堵塞、气体的生成未能及时有效的排出产生的气相堵塞.例如:Coulibaly等[17]、生贺等[18]、温春宇[19]Newman等[20]实验研究中指出, 乳化油碳源原位生物修复污染地下水时,由于乳化油在多孔介质中的滞留作用、油滴粒径大于多孔介质孔径产生的截留作用,致使乳化油在注入含水层中迁移时造成渗透性一定程度的损失.目前,关于乳化油粒径、介质颗粒大小、孔隙尺寸大小以及表面粗糙程度等对多孔介质渗透性损失的影响已得到了广泛的关注[21].然而,对于气体的生成和生物量的积累引起的渗透性变化却依然关注较少.
Hunter[5]、Long 等[22]、Borden 等[23]、Liang等[24]在乳化油原位修复含水层TCE、PCE和氯化物溶剂污染时指出,微生物的繁殖、N2、CO2等气体的生成导致含水层多孔介质堵塞,渗透性损失.为此,本文尝试从乳化油在地下水原位修复过程中产气对堵塞的影响这一角度角度出发,分别以纳米乳化油、吐温80和司盘80作为碳源开展硝酸盐降解批实验,比较纳米乳化油和两种乳化剂分别作为碳源对硝酸盐的降解效果,分析比较主要产气成分和产气量,为乳化油修复硝酸盐污染地下水过程中气体堵塞含水介质导致渗透损失提供理论基础.
1 材料与方法
1.1 实验材料
1.1.1 实验试剂 (1)纳米乳化油配制用材料:食用大豆油、吐温 80、司盘 80、去离子水.(2)微生物接种用材料:活性污泥(取自清河污水处理厂):将活性污泥在300目(孔径38µm)的滤网上过滤,取过滤液为实验所用.(3)三氮、碱度分析测试用试剂:硝酸钾、氢氧化钠、过硫酸钾、氨基磺酸、酒石酸钾钠、碳酸钠、对-氨基苯磺酰胺、N-(1-萘基)-乙二胺二盐酸盐、盐酸、磷酸、纳氏试剂.
1.1.2 实验设备 岛津UV-1800紫外可见分光光度计,上海元析仪器有限公司;JJ-1电动增力搅拌器,常州市环宇科技仪器厂;DFD-700超声波清洗器,高电电子仪器厂;HH-M6恒温水浴箱,上海赫田科学仪器有限公司;877-Titrino plus自动电位滴定仪,苏州赛恩斯仪器有限公司;GC-7980A气象色谱仪,天美科学仪器有限公司.
1.1.3 纳米乳化油的制备 本实验用纳米乳化油由课题组何宝南[21]制备而成.将大豆油、食品级乳化剂(吐温 80、司盘 80)和去离子水按照质量比为 3:3:14加入到 150mL的锥形瓶中混合,然后在电动增力搅拌器下,以 1500r/min的速率搅拌 10min,取下放入超声波清洗器中超声处理20min.超声处理结束后,利用恒温水浴箱加热至相转变温度,静置48h无破乳分层现象,即得到实验用纳米乳化油,纳米乳化纳米乳化油的理化性质如表1所示.
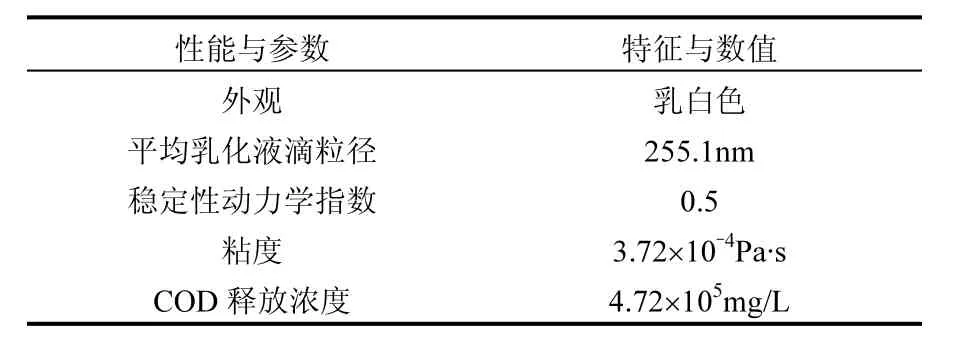
表1 纳米乳化油的理化性质Table 1 The physical and chemical properties of nano emulsified oil
1.2 实验方法
1.2.1 实验设计 共设计5组实验,每组实验均包含一个平行对照组,实验均在常温条件下(20~25℃)进行.实验前,利用锡箔纸包裹反应容器,达到避光目的.实验过程中,控制反应器溶液体积为800mL,曝氩气10min,具体实验设计如表2所示.其中,#1和#2为空白组,用于对比验证纳米乳化油(#3)、吐温80(#4)和司盘80(#5)碳源原位降解硝酸盐氮的有效性.当反应体系中硝酸盐氮的浓度降低至不再变化时,记作一个周期.待该周期结束后,通过向反应体系中添加硝酸盐溶液的方式,将反应体系硝酸盐氮提升至原始的含量水平 50mg/L,进行第二周期硝酸盐氮的降解,按此方式依次循环往复,直至碳源消耗殆尽.
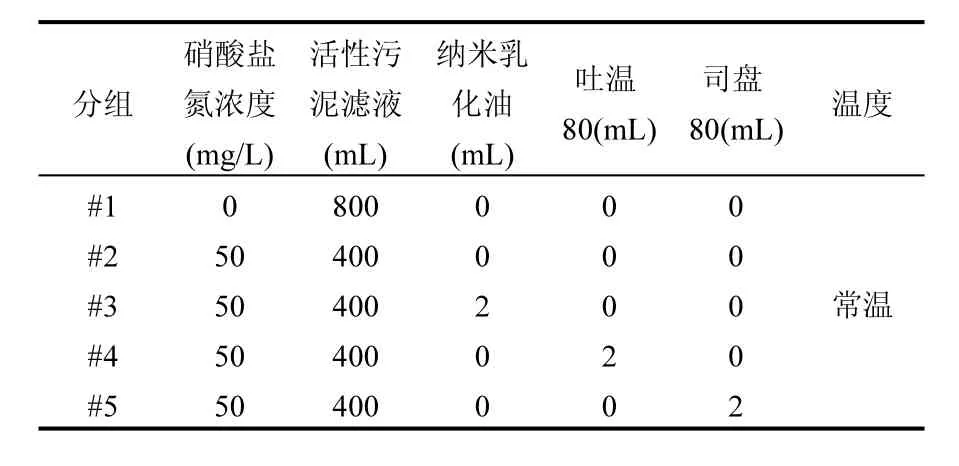
表2 各组初始加入的物质Table 2 Substances initially added in each group
#1和#2主要用于对比分析在没有碳源的条件下,活性污泥滤液对硝酸盐氮的降解效果以及降解过程中的产气情况.#3、#4、#5和#2主要用于对比分析在排除活性污泥滤液对硝酸盐氮降解的前提条件下,判定纳米乳化油、吐温80和司盘80能否作为碳源有效去除硝酸盐氮,对比不同类型碳源对硝酸盐氮的降解效果,并探究各类型碳源在降解硝酸盐氮过程中的归趋.
1.2.2 实验装置 依据瓦呼仪工作原理,设计本实验用硝酸盐氮降解与测气装置,具体实验装置如图1所示.其中,反应容器用于测定硝酸盐氮降解情况; CO2吸收瓶与U型管用于量化反应过程中的产气情况.反应容器为规格 1L的锥形瓶,装有表2设计投加物质的混合液800mL.用套针穿透的橡胶塞密封,套针用于进取样. CO2吸收瓶为规格I L的锥形瓶,装有浓度0.2mg/L的氢氧化钠溶液 1L,用于吸收碳源厌氧发酵过程中产生的CO2,从而监测和计算出反应体系 CO2的产气量.U型管由内径1.3cm、高15cm的有机玻璃管构成,侧面设置一取气口,用于分析测定反应体系产气类型,且仅在取气时用注射器穿透,U型管液位差用于记录除 CO2外反应体系中其他气体的总产气量,各反应器间通过玻璃导管密封连通.
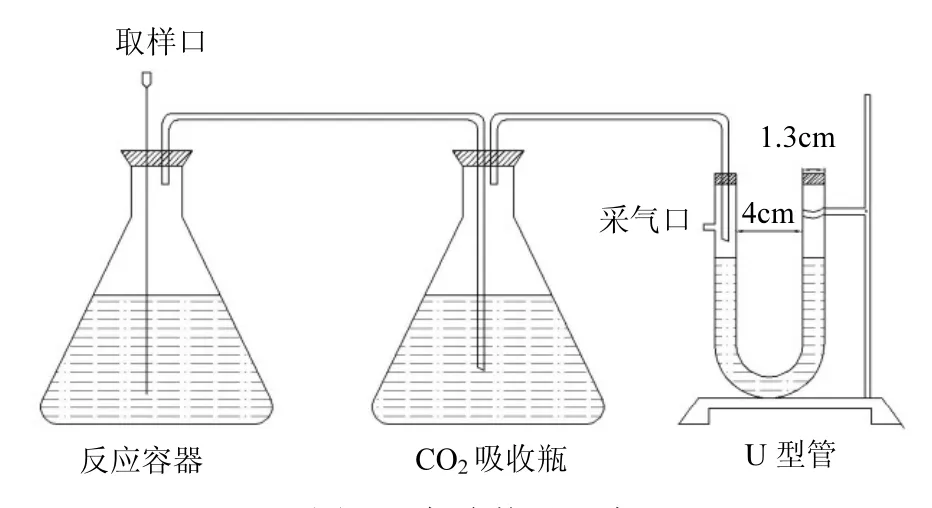
图1 实验装置示意Fig.1 Schematic diagram of experimental set-up
1.2.3 实验运行及取样分析 实验设计不同的时间间隔取样,每次取样前将套针打开,反应容器取样前先手动摇匀、混合,用一次性注射器取样15mL; CO2吸收瓶用一次性注射器取样 30mL;取气时用一次性注射器取样 30mL,每次取样结束后用套针封闭,再用封口膜封闭套针部分,以确保反应装置的封闭性.硝酸盐氮采用紫外分光光度法[25];亚硝酸盐氮采用 N-(1-萘基)-乙二胺光度法[25];氨氮采用纳氏试剂光度法[25];碱度的测试采用电位滴定法[25];气体成分使用 GC-7980A气相色谱仪测定.
1.2.4 产气量监测及计算 (1)CO2产气量:通过电位滴定法测定 CO2吸收瓶的碱度来量化反应体系CO2产气量.其原理主要依据是在酸碱指示剂条件下,标准酸溶液滴定水样至规定 pH值时的消耗量来判定.令以酚酞作指示剂时,滴定至颜色变化所消耗盐酸标准溶液的量 V1mL,以甲基橙作指示剂时盐酸标准溶液用量为(V2-V1)mL,则盐酸标准溶液总消耗量为 V2mL.根据达到两个滴定终点所消耗标准盐酸溶液用量,即可计算出 CO2吸收瓶中碳酸根和重碳酸根含量,具体计算公式如下[25]:
a. 当 V1>1/2V2时,

b. 当 V1<1/2V2时,
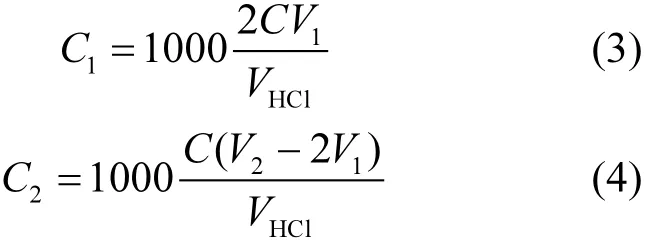
c. 当V1=0时,

式中:C1为溶液中碳酸根浓度,mol/L;C2为溶液中重碳酸根浓度;CHCl为盐酸标准溶液浓度,mol/L;VHCl为盐酸标准溶液用量,mL;V3为测样体积,25mL.
(2)U型管产气量计算方法
利用波义耳定律计算U型管内气体量:
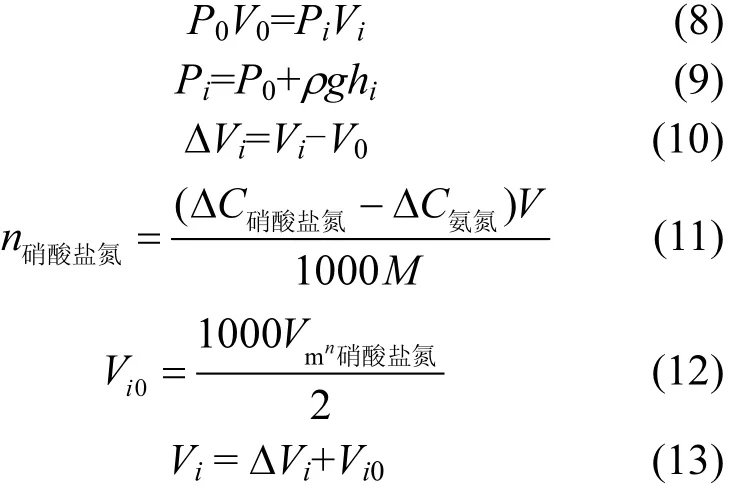
式中:P0为大气压强,1.01×105Pa;Pi为第 i次取样前实验体系的压强,Pa;V0为实验体系初始体积,mL;Vi为第i次取样前实验体系的体积,mL;hi为第i次取样前实验体系的页面差,cm;ΔVi为第i次取样前U型管变化产生的体积,mL.为第i+1次取样与第 i次取样之间硝酸盐氮的去除量,mg/L;为第i+1次取样与第i次取样之间氨氮的生成量,mg/L;V为第i次取样时反应溶液的体积,L;M 为硝酸根的摩尔质量,62g/mol;n硝酸盐氮为第i+1次取样与第i次取样之间除去转化为氨氮的那一部分硝酸盐氮,剩余硝酸盐氮的量,mol;Vi0为第i+1次取样与第i次取样之间剩余硝酸盐氮生成氮气的量,mL;Vm为物质的量体积,22.4L/mol;Vi为第i+1次取样与第i次取样之间除CO2外反应体系中其他气体的总产气量,mL.
2 结果与讨论
2.1 纳米乳化油碳源降解硝酸盐效果
2.1.1 反应体系氮素浓度变化 通过测定反应体系中三氮和总氮的浓度变化,考察纳米乳化油、吐温80和司盘80碳源在各周期内对硝酸盐氮的去除效果,各反应体系氮素浓度变化如图 2所示,实验过程中,共完成 7个周期的有效降解,图中分别用数字1~7表示.
(1)硝酸盐氮浓度变化
各反应体系中硝酸盐氮浓度变化如图 2(I)所示,其中,仅存在活性污泥滤液的#1反应体系在整个试验周期内硝酸盐氮浓度很低,为 0~1.416mg/L,一定程度上可忽略不计.与#1对比,在没有投加碳源的条件下,#2中的活性污泥滤液在前 4个周期内能够有效降解反应体系中的硝酸盐氮,但是后3个周期均不再降低,表明活性污泥对水中硝酸盐氮有一定的去除作用,但去除寿命有限,仅为4个周期,其原因是#2反应器中由于没有外加碳源,实验初期硝酸盐氮在活性污泥滤液中的微生物作用下进行反硝化反应,随着实验的进行,微生物缺少生长所需物质,开始衰亡,此时硝酸盐氮浓度不再降低.#3、#4和#5反应体系,与#2对比显示,投加碳源的反应体系在整个实验周期内硝酸盐氮浓度均有所降低,且在前4个周期内降低程度更大,表明在前4个周期内,硝酸盐氮浓度的降低是活性污泥和碳源共同作用的效果.第4周期之后,#3、#4和#5反应体系中的硝酸盐氮浓度还在降低,说明在第4周期后碳源作为活性污泥滤液中的微生物生长所需物质,促进反硝化作用,实现硝酸盐氮的降低,此时硝酸盐氮浓度的降低主要是外加碳源的作用.尽管外加碳源能够进一步实现硝酸盐氮的降解,但受碳源类型影响,硝酸盐氮降解效率存在明显差异.#3、#4和#5对比显示,实验过程中纳米乳化油碳源对硝酸盐氮的去除效果明显优于吐温80和司盘80,但在进行至约100d时,去除效率有所降低,表明纳米乳化油作为电子供体能够最大程度的促进硝酸盐氮的降解,但随着反应体系中纳米乳化油碳源的消耗,降解效率随之降低.
(2)亚硝酸浓度变化
各反应体系中亚硝酸盐氮浓度的变化如图2(II)所示,除#2反应体系在第2、3周期内亚硝氮浓度较高外,整个反应体系亚硝氮均维持在一个较低的含量水平.分析认为硝酸盐还原酶作用比亚硝酸盐还原酶作用的速率快,导致硝酸盐氮的还原率大于亚硝酸盐氮的还原速率[26-27];此外,恢复硝酸盐氮至初始含量水平时,系统初始时不太稳定,导致亚硝酸盐氮有一个逐步积累的过程,后期随着系统的稳定,亚硝酸盐氮浓度都低于检出限,并趋于稳定[27].在实验进行至100d左右时,整个反应体系中亚硝酸盐氮含量极低,基本没有积累,表明长期利用碳源强化降解地下水硝酸盐污染过程中,不会因亚硝酸盐氮的累积从而造成二次污染.

图2 不同条件下硝酸盐氮、亚硝酸盐氮和氨氮浓度随时间变化Fig.2 The concentration change of nitrate, nitrite and ammonia nitrogen with time under different conditions

(3)氨氮浓度变化
各反应体系中氨氮浓度的变化如图 2(III)所示,整个实验过程中,#1反应体系中活性污泥中的氨氮含量水平均维持在 5.88~10.16mg/L之间稳定波动.与#1对比显示,加入硝酸盐氮后,#2反应体系中氨氮含量随实验周期呈现明显的降低趋势,且小于#1反应体系中的氨氮含量水平;在第5周期之后,#3反应体系中氨氮含量显著降低,小于#1反应体系中氨氮含量水平;#4反应体系中氨氮的变化保持原有的态势;#5反应体系中氨氮含量在整个实验过程中比较稳定,略低于#1反应体系中氨氮的含量水平.
2.1.2 碳源效果评估 为了进一步研究纳米乳化油碳源效果,并与相关文献中其他碳源评估作对比,分别对各个阶段中硝酸盐氮的去除量、总去除量、去除率、总去除率和单位体积碳源去除硝酸盐氮的量进行计算,如表3所示.从上述分析可以看出,前4个周期活性污泥发挥作用,到第 5周期硝酸盐氮浓度变化的差异主要是碳源的作用.从表3可看出,从第5周期开始随着实验的进行#3~#5各组硝酸盐氮的去除率逐渐减少,去除率分别高达 99.5%、87.3%、79.8%,总去除率分别为79.5%、63.8%和68.8%,单位体积碳源去除硝酸盐氮的量分别为113.8, 91, 98.9mg/mL.对比国内外学者用不同碳源去除地下水硝酸盐效果:Hunter采用沙箱实验模拟植物油原位反应墙去除地下水中硝酸盐污染,在实验运行的30周内,硝酸盐氮的去除率为39%[15];刘菲[28]采用乳化油去除硝酸盐模拟柱实验表明,硝酸盐氮进水浓度分别为 20mg/L,50mg/L,总去除率为 73%,70%;Rocca等[29]利用零价铁和棉花作为反应介质去除地下水硝酸盐,硝酸盐氮的去除率超过 90%;Saliling等[30]以硬木屑和小麦秆作为碳源研究这两种固体碳源的反硝化效果,结果表明硝酸盐氮的去除率高达99%.对比显示,固相碳源和传统液相碳源都对地下水硝酸盐有良好的去除效果,但是具有其自身的局限性,通过将油、乳化剂和水混合制备纳米乳化油,降低注入液体的粘性、粒径,既投加方便、易于溶解,又具有固态碳源缓慢释放、易于生物降解等优点.因此,选择纳米乳化油作为原位复过程中的碳源具有一定的优势.
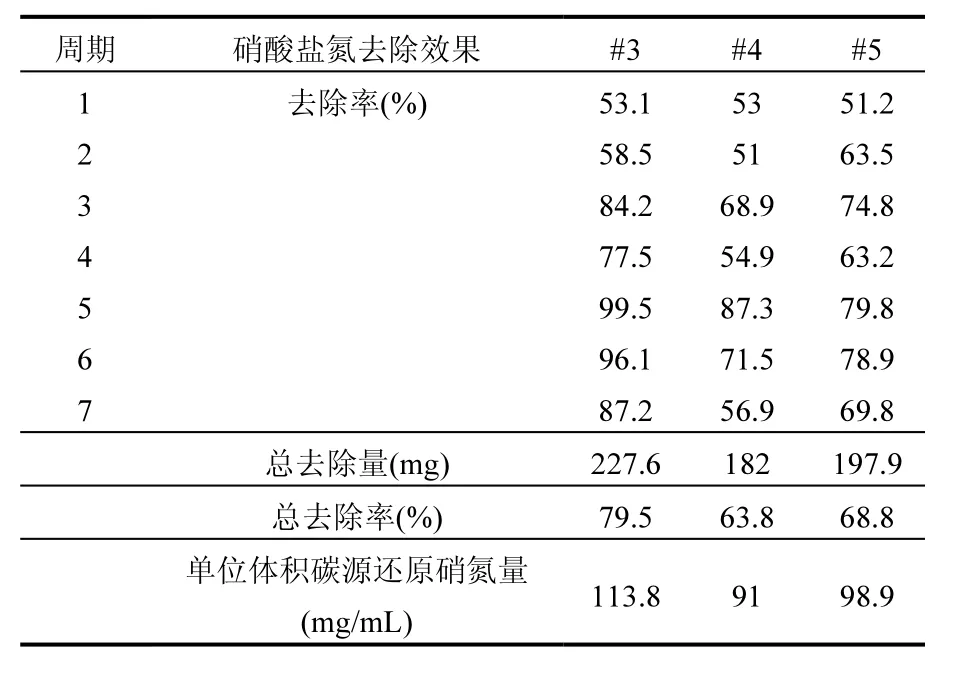
表3 不同条件下碳源效果评估分析Table 3 Assessment on effects of carbon sources under different conditions
2.2 纳米乳化油原位处理地下水硝酸盐过程中产气的变化
2.2.1 气体释放量理论计算 为了对纳米乳化油原位处理地下水硝酸盐过程中产气量有大概了解,并且与实际产气量进行对比,产气释放理论计算如下:
纳米乳化油由大豆油、吐温80、司盘80和去离子水按一定的质量比配制而成,分别计算每种组分的产气量.
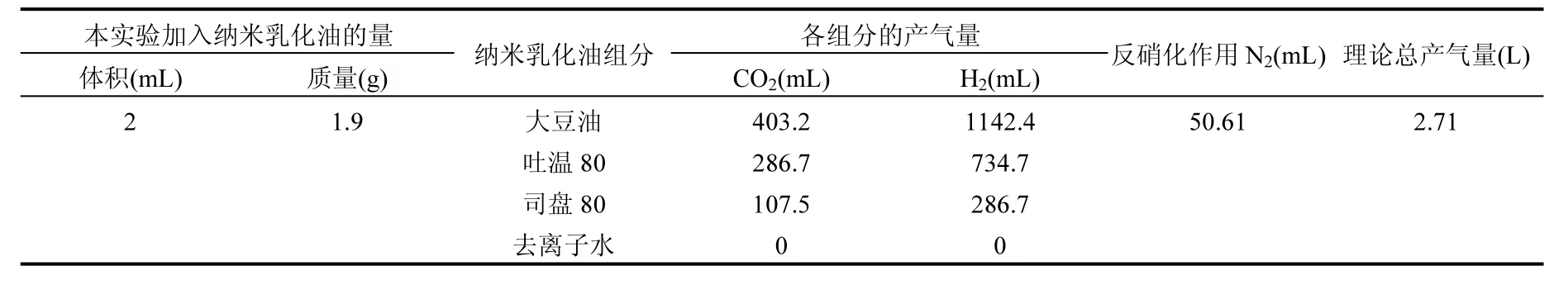
表4 气体释放量理论计算结果Table 4 Theoretical calculation results of gas production
(1)大豆油:假设在整个实验过程中油最终都发酵生成CO2和H2,有文献[31]表明大豆油厌氧发酵产生的气体如公式(14):

按照上述大豆油的分子式及反应计量关系,经计算可知:纳米乳化油中每克大豆油分别释放0.1792mol H2、0.018mol CO2,本实验过程中2mL纳米乳化油中油释放CO2和H2的理论数值分别为VCO2=403.2mL,VH2=1142.4mL.
(2)乳化剂:吐温80和司盘80分子式分别为C64H124O26、C24H44O6,假设实验过程中全部生成H2和 CO2,公式如(15)所示[31]:
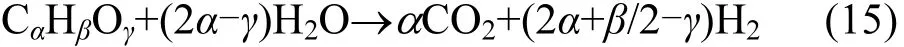
按照上述吐温80和司盘80的分子式及反应计量关系,经计算可知:本实验过程中 2mL纳米乳化油中吐温80释放的CO2和H2的理论数值分别为VCO2=286.7mL,VH2=734.7mL;司盘80释放的 CO2和 H2的理论数值分别为 VCO2=107.5mL,VH2=286.7mL.
(3)反硝化作用:实验每一周期都把硝酸盐氮浓度重新提高至50mg/L,假设50mg/L的硝酸盐氮全部反硝化转化成N2,反硝化公式如(16)所示:
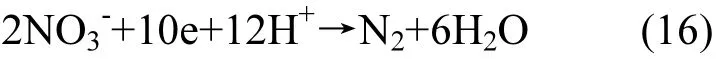
按照上述反应计量关系,经计算可知:每一周期产生N2的理论数值为7.23mL,实验总共进行7个周期,共产生N2的理论数值为50.61mL.
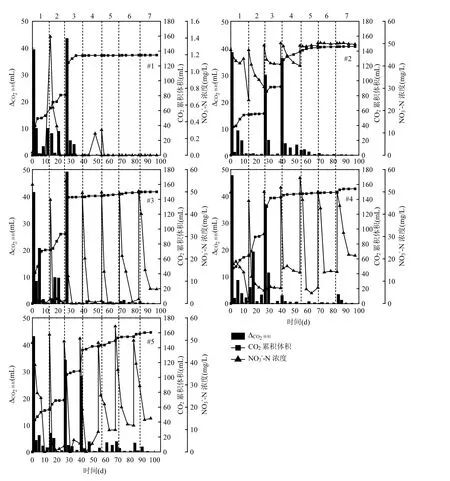
图3 不同条件下CO2产气量随时间变化Fig.3 Change of the CO2 production with time under different conditions
本课题组赵阅坤等在进行乳化油原位治理地下水中 Cr(Ⅵ)实验研究中指出,整个实验过程历时约330d,加入2mL乳化油碳源一直在发挥作用[32].依据其实验结果,假设7个周期大豆油、吐温80和司盘80全部厌氧发酵分解,其中产气中有一部分H2作为电子供体发生反硝化作用,因此应扣除 303.45mLH2.如果本实验中纳米乳化油碳源一直发挥作用,理论计算得到整个实验过程产气量约2.71L,如表4所示.
2.2.2 CO2产气量与U型管产气量 为探讨实验中各体系的产气量,分别从CO2吸收瓶和U型管两部分进行计算讨论.
(1)CO2产气量
各反应体系中 CO2产气量的变化如图 3所示,各反应体系中均有CO2产生,CO2累积产气量随实验周期趋势逐渐趋于平缓.#1、#2反应体系中没有投加碳源,CO2在逐渐积累,说明微生物自身进行新陈代谢; #3、#4、#5与#2对比显示,CO2产气量要高于#2,表明纳米乳化油、吐温80和司盘 80进行厌氧发酵产生 CO2;从#3、#4、#5柱形图可以看出,CO2产气量随着实验周期在逐渐降低,表明反应体系投加纳米乳化油、吐温80和司盘 80碳源在实验前 4个周期进行厌氧发酵产生CO2,随着碳源的不断消耗,CO2的量也随之减少.
(2)U型管产气量
各反应体系中 U型管产气量的变化如图 4所示,各反应体系中U型管均有气体累积,其中#3添加纳米乳化油反应体系U型管产气量最大,其原因是该反应体系中反硝化作用更强,释放的N2
较多.#2与#1对比显示,#1反应体系中只添加活性污泥,硝酸盐氮浓度很低,第3d后U型管液面不再发生变化,产气很少,#2反应体系中当硝酸盐氮浓度很低时,U型管产气量明显增加,表明#2反应体系发生反硝化作用, 有 N2生成;#3与#2对比发现,#3反应体系U型管累积产气量高于#2,表明投加纳米乳化油的反应体系中主要是发生反硝化作用释放N2,少量伴随纳米乳化油自身进行厌氧发酵产生的H2、CH4等气体;#4、#5与#3对比显示,#4、#5反应体系中U型管产气量低于#3,表明添加吐温80和司盘80 可以作为碳源强化降解硝酸盐氮,但是相比较之下,投加纳米乳化油反硝化作用更强,对应产气量也较大.
2.2.3 产气效果评估分析 为了进一步研究纳米乳化油碳源原位降解硝酸盐过程中产气效果,分别对 CO2产气量、U型管产气量、总产气量和产气成分进行计算分析,如表5所示.
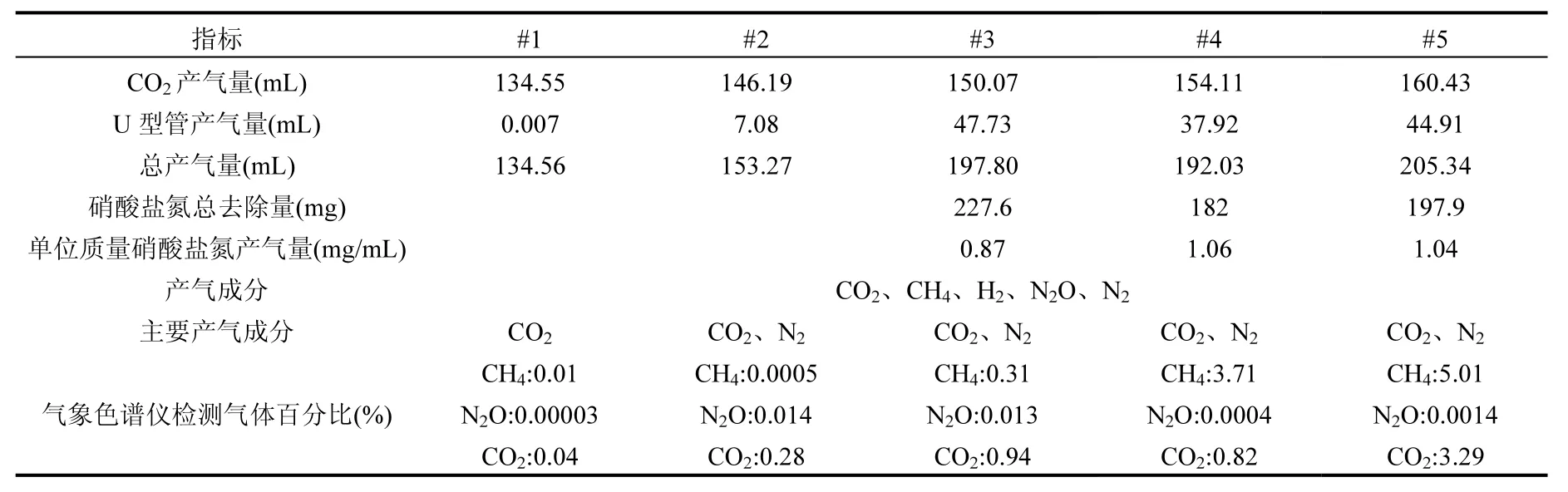
表5 不同条件下产气效果评估分析Table 5 Assessment on gas production effect under different conditions
从表5可以看出,实验约第100d时,7个周期各反应体系中均有气体的积累,其中#3纳米乳化油反应体系U型管产气量最大,为47.73mL,表明该反应体系中反硝化作用更强,释放N2较多;#5司盘80反应体系CO2产气量、总产气量最大,分别为160.43,205.34mL,说明与#3、#4对比,#5反应体系中投加2mL司盘80能较大程度地厌氧发酵产生CO2.其中各个反应体系中产气成分有CO2、CH4、N2O、H2、N2,#1反应体系主要产气成分是CO2,#2~#5反 应体系主要产气成分是CO2、N2.
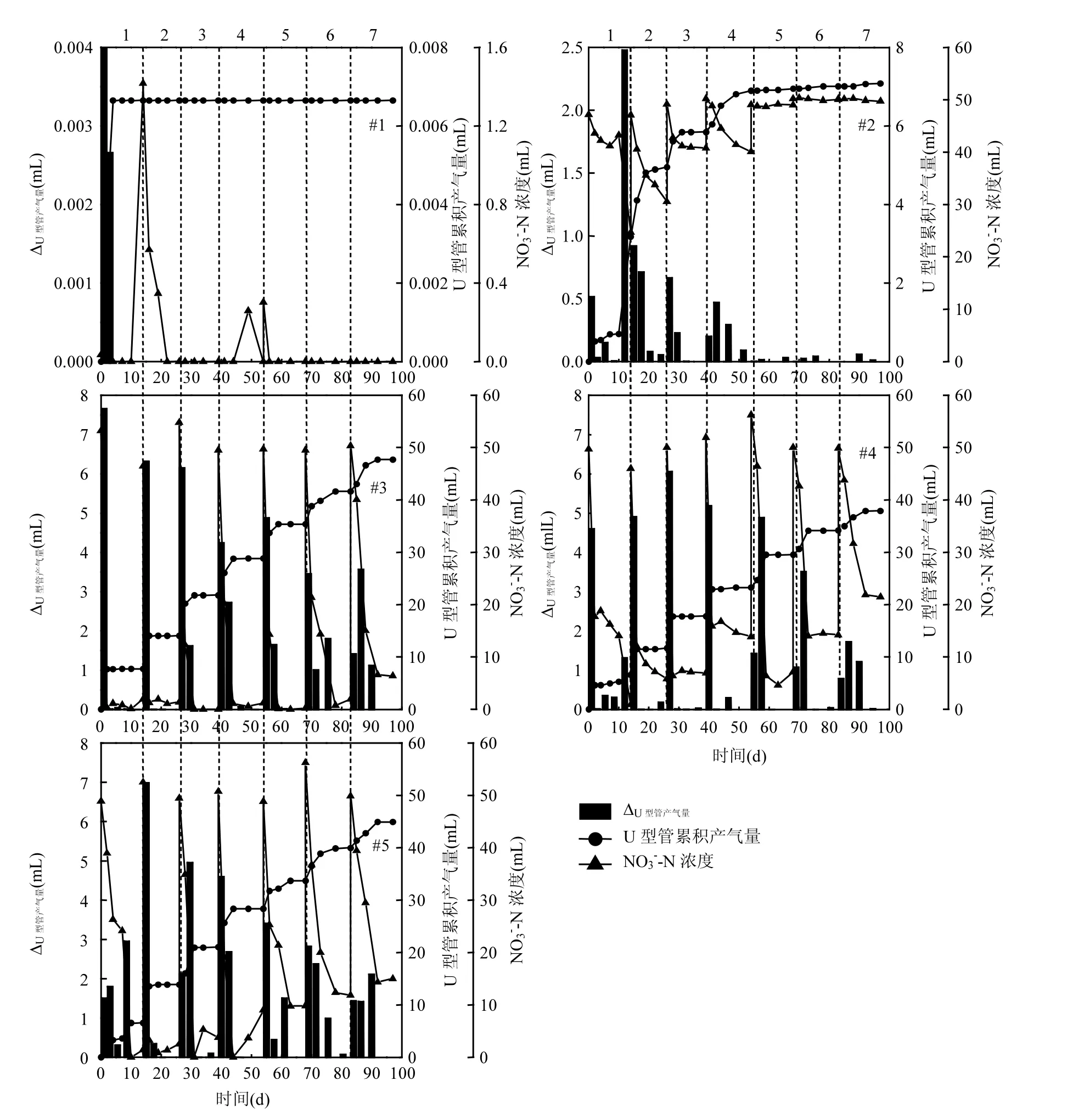
图4 不同条件下U型管产气量随时间变化Fig.4 Change of the gas production in U-shaped tube with time under different conditions
#3添加纳米乳化油反应体系中产气量与理论计算数值对比显示: CO2和N2理论计算数值分别为797.4, 50.61mL,7个周期内CO2和U型管产气量实测数值分别为 150.07, 47.73mL,实际反应过程中仅 18.8%纳米乳化油厌氧发酵产生 CO2, CO2产气量实测值与理论计算值相差比较大,一方面可能与 CO2测试方法有关,本实验是通过测定水溶液中的碳酸根和重碳酸根来表征气态 CO2,而实际水溶液中还存在游离 CO2;另一方面纳米乳化油碳源并未完全分解,即使有乙酸等副产物的产生也不会即刻被反硝化反应利用;U型管实测值几乎接近 N2理论计算值,且经过 CO2吸收瓶后,气相色谱仪检测出反应体系中 CO2、N2O、CH4气体所占百分比很小,表明纳米乳化油碳源降解硝酸盐过程中,产生的气体除 CO2外,主要是反硝化作用释放 N2.通过对纳米乳化油强化地下水硝酸盐反硝化过程中产气成分和产气量的分析评估,为产气对含水介质渗透性损失提供依据和数据支撑.
3 结论
3.1 纳米乳化油、吐温80和司盘80均能作为
碳源促进硝酸盐氮的降解,在实验的 100d内,3种反应体系中硝酸盐氮的去除率最高分别可达 99.5%、87.3%、79.8%,硝酸盐氮总去除率分别为79.5%、63.8%和68.8%,相比较之下,纳米乳化油作为碳源原位修复硝酸盐污染的去除效果最好.
3.2 实验7个周期内各反应体系中均有气体的积累,其中纳米乳化油反应体系U型管产气量最大,为47.73mL;司盘80反应体系CO2产气量、总产气量最大,分别为 160.43, 205.34mL.各个反应体系中产气成分有 CO2、CH4、N2O、H2、N2,仅添加活性污泥未添加硝酸盐的反应体系中主要产气成分是CO2,未加碳源和分别添加3种碳源的4个反应体系中主要产气成分是CO2、N2.
3.3 添加纳米乳化油反应体系中产气量与理论计算数值对比显示:CO2产气量实测值与理论计算值相差比较大,U型管实测值几乎接近N2理论计算值,且经过CO2吸收瓶后,气相色谱仪检测出反应体系中CO2、N2O、CH4气体所占百分比很小,表明纳米乳化油碳源降解硝酸盐过程中,产生的气体除CO2外,主要是反硝化作用释放N2.
[1]Borden R C. Effective distribution of emulsified edible oil for enhanced anaerobic bioremediation [J]. Journal of Contaminant Hydrology, 2007,94(1/2):1-12.
[2]Borden R C, Lieberman M T. Passive Bioremediation of Perchlorate Using Emulsified Edible Oils [J]. Journal of Contaminant Hydrology, 2009:155-175.
[3]Edible Oil Barriers for Treatment of Perchlorate Contaminated Groundwater. Federal Remediation Technology Roundtable (FRTR).Abstracts of Remediation Case Studies. 2006, http://www.frtr.gov.cn costperf. htm.
[4]Zawtocki C, Lieberman M T, Borden R C, et al. Treatment of perchlorate and 1,1,1-trichloroethane in groundwater using edible oil substrate (EOS). Proceedings of Remediation of Chlorinated and Recalcitrant Compounds-4th. Internat. Conf., Monterey, CA,,2004.
[5]Hunter W J. Use of vegetable oil in a pilot-scale denitrifying barrier [J]. Journal of Contaminant Hydrology, 2001,53:119-131.
[6]Dong J, Ding L, Chi Z F, et al. Kinetics of nitrobenzene degradation coupled to indigenous microorganism dissimilatory iron reduction stimulated by emulsified vegetable oil [J].Environmental Science (China), 2017,54:206-216.
[7]Gihring T M, Zhang G, Brandt C C, et al. A limited microbial consortium is responsible for extended bioreduction of uranium in a contaminated aquifer [J]. Applied Environmental Microbiology,2011,77(17):5955-5965.
[8]Lindow N L, Borden R C. Anaerobic bioremediatin of acid mine drainage using emulsified soybean Oil [J]. Mine Water and Environment., 2005,24:199-208.
[9]Tang G P, Waston D B, Wu W M. U(VI) Bioreduction with emulsified vegetable oil as the electron donor-model application to a field test [J]. Environmental Science & Technology,2013,47(7):3218-3225.
[10]Hiortdahl K M, Borden R C. Enhanced reductive dechlorination of tetrachloroethene dense nonaqueous phase liquid with EVO and Mg(OH)2[J]. Environmental Science & Technology, 2014,48(1):624-31.
[11]Harkness M, Fisher A. Use of emulsified vegetable oil to support bioremediation of TCE DNAPL in soil columns [J]. Journal Contaminant Hydrology, 2013,151:16-33.
[12]Wang Y, Jin L, Deshusses M A. The effects of various amendments on the biostimulation of perchlorate reduction in laboratory microcosm and flowthrough soil columns [J].Chemical Engineering Journal, 2013,232:388-396.
[13]Tang G P, Waston D B, Wu W M. U( VI) bioreduction with emulsified vegetable oil as the electron donor—microcosm tests and model development [J]. Environmental Science &Technology, 2013,47(7):3209-3217.
[14]Deng Y, Zhang P, Qin Y, et al. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation [J]. Environmental Microbiology, 2016,18(1):205-218.
[15]Zhang P, Nostrand J D V, He Z, et al. A Slow-Release Substrate Stimulates Groundwater Microbial Communities for Long-Term in Situ Cr(VI) Reduction [J]. Environmental Science &Technology, 2015,49(21):12922-12931.
[16]Watson D B, Wu W M, Mehlhom T, et al. In situ bioremediation of uranium with emulsified vegetable oil as the electron donor [J].Environmental Science & Technology, 2013,47(12):6440-6448.
[17]Coulibaly K M, Borden R.C. Impact of edible oil injection on the permeability of aquifer sands [J]. Journal of Contaminant Hydrology, 2004,71(1-4):219-237.
[18]生 贺.乳化油反应带强化修复 Cr(Ⅵ)污染地下水研究 [D].吉林:吉林大学, 2015.
[19]温春宇.乳化纳米铁降解硝基苯污染地下水的研究 [D]. 吉林:吉林大学, 2014.
[20]Newman W A, Pelle R C. Enhanced anaerobic bioremediation of chlorinated solvents utilizing vegetable oil emulsions [J].Remediation Journal, 2006,16(3):109-122.
[21]何宝南.纳米乳化油制备及其在多孔介质中的迁移释放模拟实验 [D]. 北京:中国地质大学(北京), 2017.
[22]Long C M, Borden R C. Enhanced reductive dechlorination in columns treated with edible oil emulsion [J]. Journal Contaminant Hydrology, 2006,87(1/2):54-72.
[23]Borden R C. Concurrent bioremediation of perchlorate and 1,1,1-trichloroethane in an emulsified oil barrier [J]. Journal of Contaminant Hydrology, 2007,94(1/2):13-33.
[24]Liang S H, Kuo Y C, Chen S H et al. Development of a slow polycolloid-releasing substrate (SPRS) biobarrier to remediate TCE-contaminated aquifers [J]. Journal Hazardous Materials,2013,254-255:107-15.
[25]国家环境保护总局.水和废水监测分析方法 [M]. 4版.北京:中国环境科学出版社, 2002.
[26]董 萌.地下水硝酸盐污染的原位修复试验研究 [D]. 邯郸:河北工程大学, 2015.
[27]阴 娟.地下水硝酸盐污染原位修复实验研究 [D]. 合肥:合肥工业大学, 2011.
[28]刘 菲.地下水硝酸盐乳化油原位修复模拟柱实验研究 [D].北京:中国地质大学(北京), 2014.
[29]Rocca C D, Belgiomo V, Meric S. Heterotrophic/autotrophic denitrification (HAD) of drinking water: prospective use for permeable reactive barrier [J]. Desalination, 2007,210(1-3):194-204.
[30]Saliling W, Jones B, Westerman P W et al. Wood chips and wheat straw as alternative biofilter media for denitrification reactors treating aquaculture and other wastewaters with high nitrate concentrations [J]. Aquacultural Engineering, 2007,37(3):222-233.
[31]Protocol for Enhanced In Situ Bioremediation Using Emulsified Edible Oil [R]. 2006.
[32]赵阅坤,何江涛,王 磊,等.乳化油碳源原位治理地下水中Cr(Ⅵ)静态批试验研究 [J]. 环境工程, 2016,(a1):220-225.
