适用于二代测序技术的细菌DNA提取方法的比较研究
2018-06-20岳巧云刘德星陈利伟邱德义何秋星
陈 佩 岳巧云 刘德星 陈利伟 邱德义* 何秋星
(1.广东药科大学,广东广州 510006;2.中山出入境检验检疫局)
1 前言
细菌是非常重要的一类生物种群,与人类健康和社会发展息息相关[1],一直是检验检疫领域的重点检测对象,尤其在国境卫生检疫中,口岸医学媒介生物携带病原体的检测对于防止虫媒传染病通过国境口岸输入、输出发挥着重要作用,而细菌作为病原体的一种,如何快速、准确、全面地将其鉴定检测出,是防控的基础和前提[2]。传统的细菌研究方法主要分为两类,一是基于对细菌分离培养的生化鉴定方法,但绝大多数(99%以上)细菌无法在现有实验室条件下进行培养[3-4];二是基于特异引物/探针/抗体的方法,均依赖于已知细菌的基因组序列,对于检测未知的病原体存在很大的局限性[5]。
宏基因组学的兴起以及二代测序技术的迅速发展,可同时对上百份样本进行高通量核酸分子测序[6],不依赖于细菌的分离培养,直接对样品中所有的细菌进行研究,进而可以全面地对所有细菌进行检测分析[7]。目前二代测序技术已广泛运用于多个领域不同样品中携带细菌的检测分析,包括土壤[8]、人体肠道[9]、水体[10]、空气[11]、媒介生物[12-13]等,然而从最初的细菌核酸提取到后期的数据分析,二代测序技术的结果质量会受到多方面因素的影响,如细菌DNA的提取方法[14-15],16S rRNA高变区的选择[16-17],测序文库的构建,测序平台和策略的选择[5]等。在实际检验检疫应用中,基于携带病原体的宿主不同,待检样品中细菌物种多样性丰富,细菌DNA的提取作为后续扩增、测序和分析的基础,如何高效率、全面地提取出样品中的细菌DNA,对于结果的影响尤为突出。目前细菌DNA的提取方法多种多样,原理各不相同,各有其优缺点[18]。本研究比较了细菌的6种DNA提取方法,对混合细菌样本进行DNA提取,并特异性地扩增16S rRNA的V3-V4高变区基因,构建文库并分析测序结果中各种细菌的含量和相对分布,从而得到一种高效、适用范围广的细菌核酸提取方法。
2 材料与方法
2.1 材料
2.1.1 实验样品
实验菌株共10种,包含5种革兰氏阳性菌,分别为金黄色葡萄球菌(Staphylococcus aureus)、蜡样芽孢杆菌 (Bacillus cereus)、 粪肠球菌(Streptococcus faecalis)、马红球菌(Rhodococcus equi)、英诺克李斯特氏菌(Listeria innocua),以及5种革兰氏阴性菌,分别是肺炎克雷伯菌(Klebsiella pneumoniae)、绿脓杆菌 (Pseudomonas aeruginosa)、 大肠埃希菌(Escherichia coli)、肠沙门菌(Salmonella enterica)、弗氏志贺菌(Shigella flexneri)。以上菌株均为普通琼脂斜面保存的标准菌株,由中山出入境检验检疫局技术中心提供。
2.1.2 主要试剂与仪器
主要试剂:MagPure Bacterial DNA KF Kit(Megen生物);TIANamp Bacteria DNA Kit(天根生化科技有限公司);GCM107营养琼脂(北京陆桥技术有限责任公司);溶菌酶(源叶生物 R21037);蛋白胨(AOBOX);牛肉膏 (广东环凯微生物科技有限公司)。
仪器:全波长酶标仪(Thermo ScientificTMMulti skanTMGo);超声波破碎仪(Sonics Vibra-Cell);核酸提取仪(Thermo KingFisher Duo Prime);电泳仪(北京六一生物科技有限公司);凝胶成像仪(UVltec);PCR 仪(Life Technologies applied biosystems);恒温摇床(北京五洲东方科技发展有限公司)。
2.2 方法
2.2.1 单一菌种的培养及分子鉴定
2.2.1.1 菌种培养
采用分区划线法分别将10种菌株接种于营养琼脂培养基上,37℃培养48 h。随机挑取3个单菌落分别接种于牛肉膏蛋白胨液体培养基中,于37℃、150 r/min恒温摇床培养箱中培养24 h,得到细菌菌液,保存于4℃冰箱备用。
2.2.1.2 细菌DNA提取
分别取10种细菌菌液1 mL置于1.5 mL离心管中,用核酸提取仪提取DNA,按照MagPure Bacterial DNA KF Kit说明书进行样品前处理,得到样品消化液之后,采用核酸提取仪中的MagPure_Bacterial_DNA_Duo程序提取DNA。
2.2.1.3 PCR扩增
扩增细菌样本16S rRNA基因,引物序列为:正向引物 27F(5′-AGAGTTTGATCCTGGCTCAG-3′)和反向引物 1492R(5′-TACGGCTACCTTGTTACGA CTT-3′)[19],PCR 体系为 50 μL:10 倍 PCR 缓冲液 5 μL;正向引物(20 μmol/L)和反向引物(20 μmol/L)各 2 μL;dNTP (10 μmol/L) 2 μL;Ex-Taq 1 μL;模板DNA 3 μL;无菌水定容至50 μL。PCR反应条件 :95℃ 变 性 5 min;94℃ 、40 s,50℃ 、30 s,72℃ 、2 min,35个循环;72℃延伸10 min。PCR反应结束后,PCR产物经电泳检测(100 V电压,340 mA电流,电泳缓冲液为1×TAE,电泳25 min),凝胶系统成像并记录。
2.2.1.4 测序及同源性分析
采用PCR产物直接测序,将PCR产物送交上海立菲生物科技有限公司测序,测序引物同扩增引物。将测序所得序列去除两端引物部分,并与Gen-Bank中相关序列进行同源性分析。利用Maga 6.0软件中的Test Maximum Likelihood tree,对10种细菌样品的16S rDNA基因序列进行ML进化树的分析。
2.2.2 制备混合菌液
以浇注平板培养法进行细菌计数,将10种菌液梯度稀释至 10-4、10-5、10-6、10-7、10-8cfu/mL 共 5 个浓度梯后,分别吸取每个梯度的混匀菌液0.2 mL于无菌平皿中,及时倒入15~20 mL冷却至46℃的营养琼脂培养基,并转动平板混匀,待平板凝固后,37℃倒置培养48 h;每个浓度梯度设置两个平行。按照GB 4789.2—2010统计菌落数,并计算菌液浓度。根据细菌计数的结果,分别取浓度数量级为108cfu/mL的10种细菌样本的菌液等量混匀,得到浓度数量级为107cfu/mL的混合菌液备用。
2.2.3 混合细菌的DNA提取
采用6种不同的方法分别提取混合菌液的DNA,每组设置3个平行样,提取出的DNA作为模板DNA于-20℃冰箱保存备用。6种提取方法如下。
2.2.3.1 磁珠法
取1 mL混合菌液于1.5 mL离心管中作为实验样品,具体步骤同2.2.1.2。其提取原理是基于高结合力的磁珠粒子的纯化方式,在高浓度的离子化合剂条件下,磁珠可通过氢键和静电作用吸附核酸,而蛋白质或其他杂质不被吸附,吸附了核酸的磁珠经洗脱去除蛋白和盐,然后用ddH2O洗脱最终得到的DNA溶液。
2.2.3.2 热裂解法
取1 mL混合菌液于1.5 mL离心管中,95℃热裂解10 min,13 000 g离心2 min收集上清至1.5 mL离心管中。
2.2.3.3 超声波法
取1 mL混合菌液于5 mL养菌管中,置于冰盒上超声间歇处理5 min(超声2 s,间隔5 s,功率20%)[20],超声处理完后,13 000 g离心 2 min收集上清至1.5 mL离心管中。
2.2.3.4 试剂盒法(溶菌酶处理)
取1 mL混合菌液于1.5 mL离心管中作为实验样品,按照TIANamp Bacteria DNA Kit说明书提取细菌DNA。
2.2.3.5 试剂盒法(无溶菌酶处理)
除了在样品消化过程中不加入溶菌酶,略过步骤2,其余操作步骤同2.2.3.4。
2.2.3.6 超声+热裂解法
取1 mL混合菌液于5 mL养菌管中,置于冰盒上超声间歇处理5min(超声2s,间隔5s,功率20%),超声处理完后,95℃热裂解10 min,13 000 g离心2 min收集上清至1.5 mL离心管中。
2.2.4 混合菌液DNA的PCR扩增
分别以6种方法提取出的混合菌液DNA为模板,扩增其16S rRNA的V3-V4高变区基因。扩增引物为:正向引物 341F(5′-ACTCCTACGGGAGGCA GCAG-3′)和反向引物 806R(5′-GGACTACHVGGG TWTCTAAT-3′)[21],PCR 体系为 50 μL:2×PCR 缓冲液 25 μL;正向引物(20 μmol/L)和反向引物(20 μmol/L)各 1 μL;dNTP(10 μmol/L) 2.5 μL;KOD FX Neo 1μL;模板 DNA 3μL;无菌水定容至50μL。PCR反 应 条 件 :94℃ 变 性 2 min;98℃ 、10 s,50℃ 、30 s,68℃、30 s,30个循环;68℃延伸 10 min。PCR 反应结束后,PCR经产物电泳检测(100 V电压,340 mA电流,电泳缓冲液为 1×TAE,电泳 25 min),凝胶系统成像并记录。
2.2.5 二代测序及数据处理
PCR产物直接送交华大基因科技有限公司进行测序,采用Illumina HiSeqTM2000测序平台,测序策略为PE300。测序流程主要为:先对样品进行检测,DNA总量不低于1.5 μg即可满足建库测序要求;然后回收目的Amplicon片段,用T4 DNA Polymerase、Klenow DNA Polymerase和T4 PNK 将打断形成的粘性末端修复成平末端,再通过3'端加碱基“A”,使得DNA片段能与3'端带有“T”碱基的特殊接头连接;最后,用合格的文库进行cluster制备和测序。下机数据经过数据过滤,滤除低质量的reads后,通过reads之间的overlap关系拼接成Tags,拼接的Tags经过优化后,在97%相似度下将其聚类为用于物种分类的OTU(Operational Taxonomic Units)。
2.2.6 不同提取方法的比较
对于6种提取方法主要从两方面进行评价,一方面,提取出的混合细菌DNA是否满足二代测序建库的要求,包括:(1)PCR产物的电泳条带是否清晰明亮;(2)PCR产物中目的DNA总量不低于1.5 μg。另一方面,二代测序结果是否能反应样品的真实情况,包括:OTU数、细菌种类的鉴定、不同细菌的相对含量分布情况。测序结果中OTU的丰度说明了样品的物种丰富程度,通过统计测序结果中OTU的数量以及每个OTU所对应的序列数,可得到样品中细菌种数及不同细菌的相对含量,然后将测序结果与样品的实际情况进行比较,从而分析不同方法之间细菌种类鉴定和相对含量分布准确性的差异。
3 结果
3.1 单一菌种的鉴定
凝胶电泳成像后,扩增16S rRNA基因的PCR产物均有长度约为1 500 bp的条带。将测序所得序列去除两端引物部分,并与GenBank中相关序列进行同源性分析,结果详见表1,10种细菌均能被准确鉴定到种的水平,表明了实验菌种的准确性。

表1 10种细菌样本的鉴定结果
将10种细菌的16S rRNA及其V3-V4高变区基因序列分别进行ML树分析 (图1),16S rRNA基因序列进化树表明10种细菌的进化距离较远,运用16S rRNA基因序列能将其准确地划分到不同的种;而在16S rRNA V3-V4高变区基因序列进化树中,仅大肠埃希氏菌和弗氏志贺菌未被区分开。
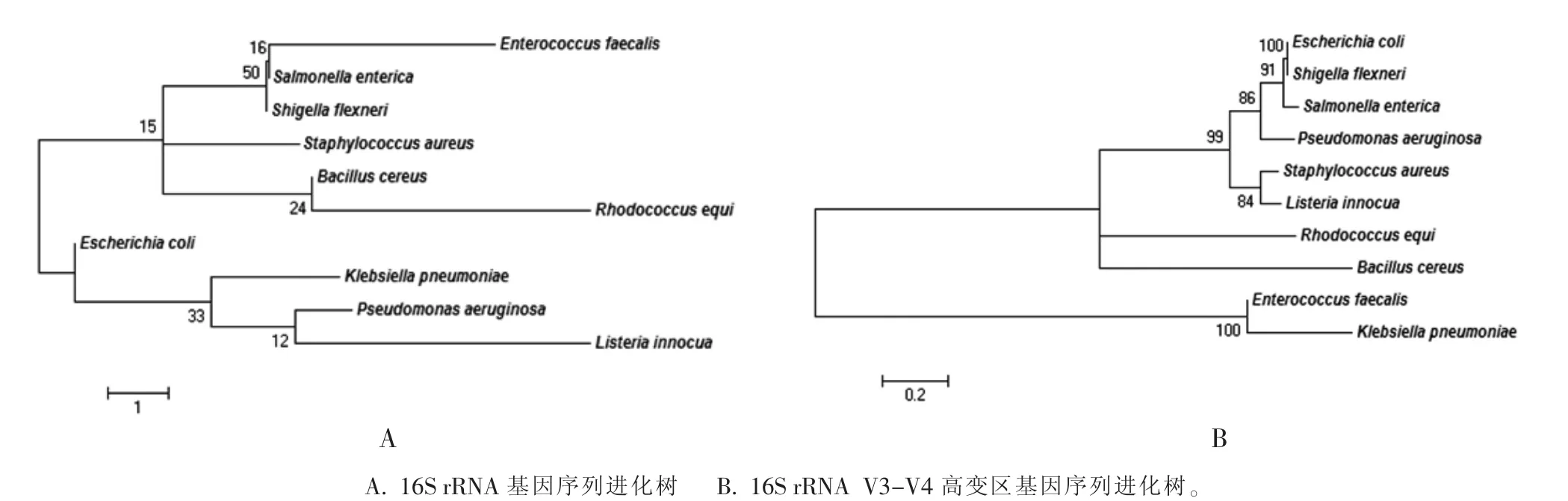
图1 10种细菌的进化树分析图
3.2 6种方法中混合细菌的鉴定
3.2.1 混合细菌DNA的浓度、纯度及PCR结果
由表2可知,6种方法中,磁珠法和试剂盒法(溶菌酶处理)提取出的DNA浓度较低,均值低于10 ng/μL,其余4种方法的浓度相近,均值大于40 ng/μL;热裂解法提取出的DNA纯度最低,其余5种方法纯度均大于1.6,其中试剂盒法(溶菌酶处理)的纯度最高。
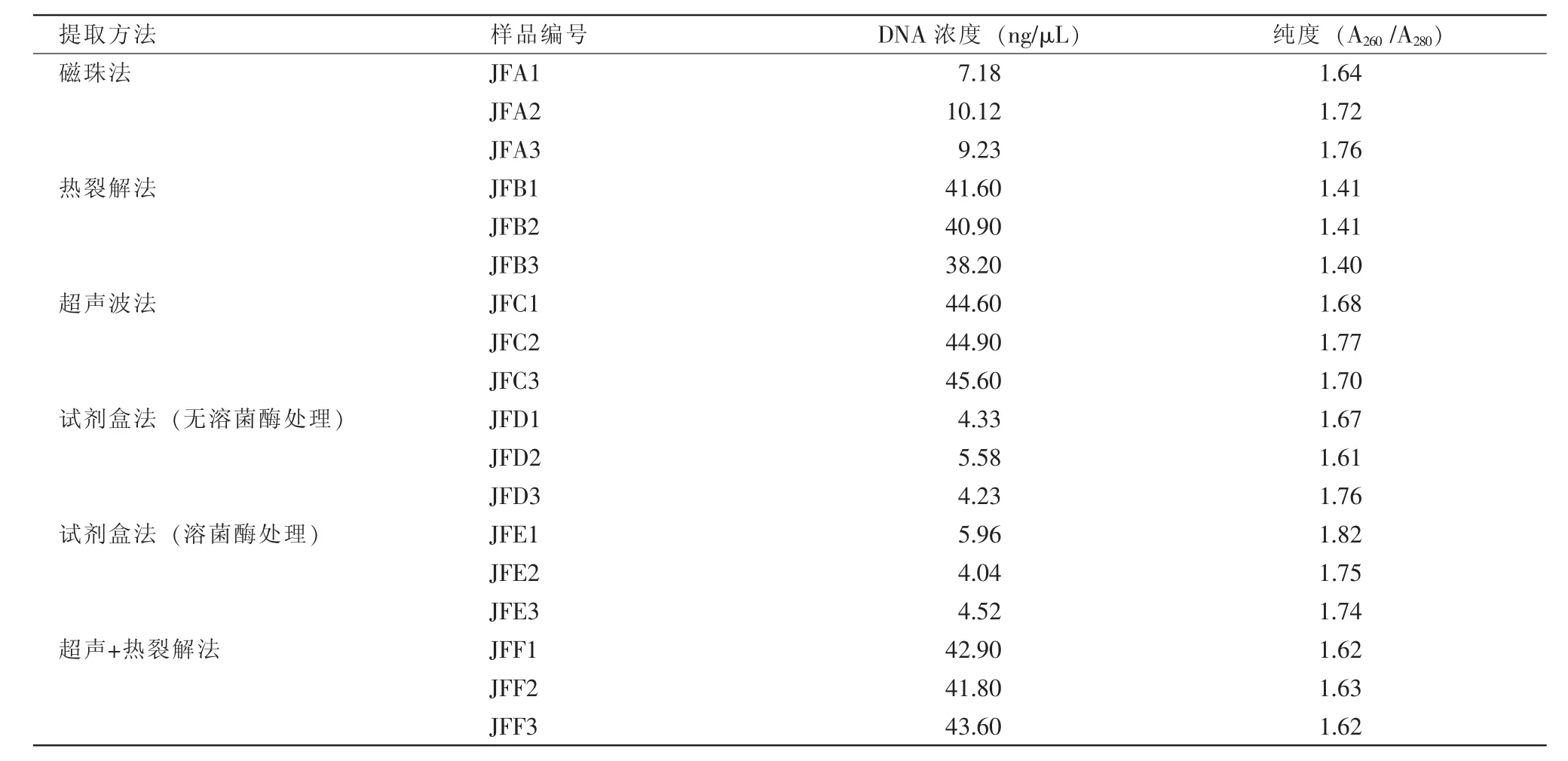
表2 6种提取方法所得细菌DNA的浓度及纯度测定结果
6种方法提取出的混合细菌DNA浓度和纯度各不相同,但经PCR扩增后,凝胶成像见图2,均有长度约为550 bp的明亮条带,且其PCR产物中目的片段的总量均不低于1.5 mg,满足建库测序的要求。
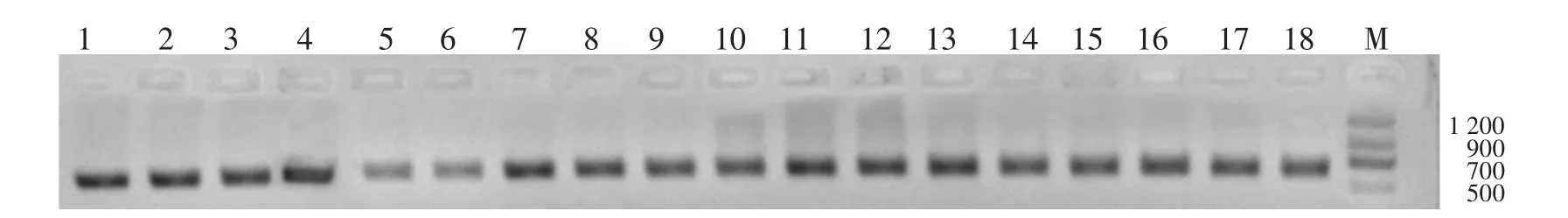
图2 混合菌液DNA的PCR产物凝胶电泳图
3.2.2 二代测序结果
3.2.2.1 OTU丰度分析
混合菌液样本中共包含10种细菌,理想的测序结果应该是将所有序列聚类为10种OTU,并且对OTU进行物种注释能准确地鉴定出样品中的10种细菌。磁珠法和试剂盒法(溶菌酶处理)测序结果的OTU数均为9,无杂质OTU,其余4种方法的OTU数均大于10,其中超声法的OTU均数为15,试剂盒法(无溶菌酶处理)为12,热裂解法和超声+热裂解法的OTU数均超过了30。表明通过磁珠法和试剂盒法(溶菌酶处理)提取细菌DNA,其测序结果中OTU的数目与样品的真实数目最为接近。
3.2.2.2 菌种种类鉴定
由表3可知,6种方法的测序结果中均只能将5种细菌鉴定到种的水平,分别为肠沙门氏菌、大肠埃希氏菌、马红球菌、蜡样芽孢杆菌和金黄色葡萄球菌,将4种细菌鉴定到属的水平,分别为克雷伯氏菌属、李斯特氏菌属、肠球菌属和假单胞菌属,而弗氏志贺氏菌均未被鉴定出。
3.2.2.3 菌种相对数量
混合菌液样品中10种细菌均是等量加入,而在测序结果中,同一方法不同菌种的相对数量分布各不相同,马红球菌、金黄色葡萄球菌和李斯特菌属的量在磁珠法、试剂盒法(溶菌酶处理)和超声+热裂解法中明显低于平均值(10%);热裂解法中大肠埃希菌和假单胞菌属的量,以及超声法中假单胞菌属的量远高于平均值,而这两种方法中其余菌种的量均较低;试剂盒法(无溶菌酶处理)中除了假单胞菌属远高于平均值以及大肠埃希菌和克雷伯菌属的量接近平均值,其余菌种的量均低于平均值。不同方法之间,同一菌种的分布也不相同;马红球菌和李斯特菌属在试剂盒法(溶菌酶处理)中提取得率最高,金黄色葡萄球菌在试剂盒法(无溶菌酶处理)中最高,但这3种菌在6种方法中提取率均低于平均值;蜡样芽孢杆菌和肠球菌属的量仅在磁珠法和试剂盒法(溶菌酶处理)中高于平均值;而大肠埃希菌除了在超声法中得率低于平均值,在其余5种方法中均较高;假单胞菌属在6种方法中的得率均较高;肠沙门菌和克雷伯菌属的量在热裂解法和超声法中均较低。
对6组测序结果中样品菌种的相对分布进行统计,计算样品菌种的序列数占总序列数的比例(表3),绘制柱状图,图3直观地反映了6种方法中样品菌种的相对分布情况。由图可观察到磁珠法和试剂盒法(溶菌酶处理)中各种细菌的含量分布相对均匀,最为接近样品中细菌含量分布的真实情况。
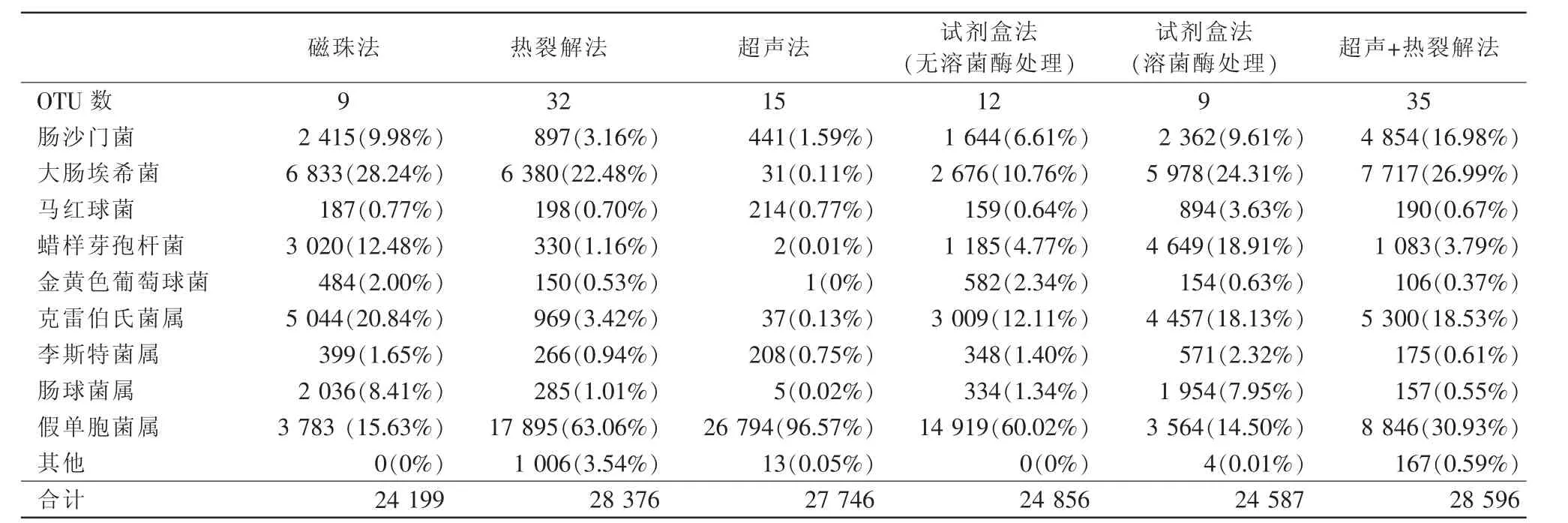
表3 6种方法测序结果中不同细菌的序列条数分布

图3 6种方法中各种细菌数据量的相对分布
4 讨论
结果表明,6种提取方法对于二代测序结果的影响主要体现在OTU丰度和菌种相对数量上,磁珠法和试剂盒法(溶菌酶处理)的OTU数最接近真实数量,无杂质OTU;而超声法和试剂盒法(无溶菌酶处理)比真实数量稍多,热裂解法和超声+热裂解法则远远超过了真实数量;说明磁珠法和试剂盒法(溶菌酶处理)测序结果较为准确,其他4种提取方法对于OTU丰度有明显影响,从而导致测序结果与真实结果相差较大。6种方法的二代测序结果中,菌种相对数量分布结果均不准确,不同的提取方法对于细菌DNA的提取影响显著,在同一方法中,不同细菌的提取效果差别很大,磁珠法和试剂盒法(溶菌酶处理)的结果中各种菌的分布情况相对均匀,最能反映样品中各种细菌含量的真实情况;对比试剂盒法(无溶菌酶处理)和试剂盒法(溶菌酶处理)的结果可知,可能是因为加入溶菌酶处理能减小细菌提取过程中所造成的结果偏倚;比较超声+热裂解法、热裂解法和超声法可知,超声+热裂解法相较于单独的热裂解法和超声法其结果均匀度有所提高。对同一种细菌使用不同方法的提取效果差别也很大,在加入溶菌酶的磁珠法和试剂盒法中,蜡样芽孢杆菌和大肠埃希氏菌的数据量远高于另外4种不加溶菌酶的方法,说明溶菌酶处理对于样本中蜡样芽孢杆菌和大肠埃希氏菌的检测至关重要。
本研究中,混合菌液样品共加入10种细菌,特异性扩增细菌16S rRNA V3-V4高变区基因用于二代测序分析,而6种方法均未将弗氏志贺菌鉴定出来。弗氏志贺菌和大肠埃希菌属于2个不同的属,分别为志贺菌属和埃希菌属,图1的ML进化树表明了用16S rRNA基因可以准确将这2种细菌区分开,但由于弗氏志贺菌的V3-V4高变区基因序列和大肠埃希菌的相似度为100%,无碱基差异,因此导致二代测序结果中未将2种细菌分开,可能将弗氏志贺菌都聚类为了大肠埃希菌,而这一结果也进一步验证了扩增16S rRNA不同高变区基因会对细菌样品构成的分析结果产生明显的影响[16-17]。
5 结论
综合考虑6种方法对二代测序结果的影响,磁珠法和试剂盒法(溶菌酶处理)用于提取混合细菌基因组DNA的效果最好,但其结果并不是十分理想,可能将多种方法结合使用会有更好的效果。鉴于传统细菌鉴定检测的不足,二代测序作为高通量快速检测技术,给全面快速地检测样品,尤其是医学媒介生物携带细菌病原体的情况提供了可能,但二代测序结果质量的影响因素较多,仍需后续更全面深入的研究和优化。
[1]Rasko D A,Sperandio V.Anti-virulence strategies to combat bacteria-mediated disease[J].Nature Reviews Drug Discovery,2010,9(2):117.
[2]孙立新,丁永键,朱临,等.江苏口岸输入性蝇类携带细菌监测研究[J]. 中华卫生杀虫药械,2011,(3):190-194.
[3]Kaeberlein T,Lewis K,Epstein S S.Isolating"Uncultivable"Microorganisms in Pure Culture in a Simulated Natural Environment[J].Science,2002,296(5570):1127.
[4]StreitW R,SchmitzR A.Metagenomics-the key to the uncultured microbes.[J].Current Opinion in Microbiology,2004,7(5):492-498.
[5]李定辰.基于高通量测序平台的未知病原微生物检测系统[D].北京:中国人民解放军军事医学科学院,2016.
[6]Mardis E R.Next-generation DNA sequencing methods[J].Annual Review of Genomics&Human Genetics,2008,9(1):387.
[7]Chaysavanh M,Chapple C E,Lionel F,et al.A comparison of random sequence reads versus 16S rDNA sequences for estimating the biodiversity of a metagenomic library[J].Nucleic Acids Research,2008,36(16):5180.
[8]Vasileiadis S,Puglisi E,Arena M,et al.Soil Bacterial Diversity Screening Using Single 16S rRNA Gene V Regions Coupled with Multi-Million Read Generating Sequencing Technologies[J].Plos One,2012,7(8):e42671.
[9]Qin J,Li R,Raes J,et al.A human gut microbial gene catalogue established by metagenomic sequencing[J].Nature,2010,464(7285):59-65.
[10]Tyson G W,Chapman J,Hugenholtz P,et al.Community structure and metabolism through reconstruction of microbial genomes from the environment[J].Nature,2004,428(6978):37-43.
[11]Hospodsky D,Qian J,Nazaroff W W,et al.Human occupancy as a source of indoor airborne bacteria.[J].Plos One,2012,7(4):e34867.
[12]韩娜,张琳,张雯,等.长角血蜱细菌群落结构及多样性研究[J].中国媒介生物学及控制杂志,2016,27(5):426-431.
[13]陈健,岳巧云,邱德义,等.二代测序技术检测大头金蝇携带细菌的研究[J]. 中国媒介生物学及控制杂志,2017,28(2):124-130.
[14]温崇庆,何瑶瑶,薛明,等.高通量测序分析DNA提取引起的对虾肠道菌群结构偏差[J]. 微生物学报,2016,56(1):130-142.
[15]Kennedy N A,Walker A W,Berry S H,et al.The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing[J].Plos One,2014,9(2):e88982.
[16]Pidiyar V J,Jangid K,Patole M S,et al.Studies on cultured and uncultured microbiota of wild culex quinquefasciatus mosquito midgut based on 16s ribosomal RNA gene analysis[J].American Journal of Tropical Medicine& Hygiene,2004,70(6):597-603.
[17]张军毅,朱冰川,徐超,等.基于分子标记的宏基因组16S rRNA基因高变区选择策略[J].应用生态学报,2015,26(11):3545-3553.
[18]郝敏,刘占英,杨天舒,等.细菌基因组DNA提取方法概述[J].生物学通报,2014,49(3):4-6.
[19]Moreno C,Romero J,Espejo R T.Polymorphism in repeated 16S rRNA genes is a common property of type strains and environmental isolates of the genus Vibrio[J].Microbiology,2002,148(4):1233-9.
[20]王伟,王玉琢,舒鹏,等.16S核糖体DNA宏基因组测序中细菌核酸提取方法的比较研究[J].生物技术通讯,2015,(4):551-555.
[21]Takahashi S,Tomita J,Nishioka K,et al.Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing[J].Plos One,2014,9(8):e105592.
