空腹胰岛素水平对高尿酸血症病人痛风发作的预测价值
2018-06-19
(青岛大学附属医院内分泌与代谢性疾病科,山东 青岛 266003)
痛风是由于血液中尿酸水平升高所致。尿酸水平较高时导致尿酸盐结晶析出,晶体会沉积在关节、肌腱和周围组织中[1]。目前认为,胰岛素敏感组织(肌肉、脂肪组织和肝脏等)对胰岛素的不敏感会加重慢性、低水平炎症。炎症被触发后可以削弱胰岛素的作用,胰岛素的作用被削弱出现胰岛素抵抗又会增强炎症的反应性,两者互相作用和影响,形成了一种恶性循环。炎症反应与胰岛素作用之间关系复杂。临床实践中发现许多合并高尿酸血症和肥胖的糖尿病病人,在胰岛素治疗后,容易发生痛风,提示胰岛素升高与痛风发作的诱发可能存在一定的关系。本研究旨在探讨胰岛素水平与痛风发作之间的关系,以及其可能诱发痛风的切点,以进一步了解痛风的发病机制,指导在治疗合并高尿酸血症或肥胖的糖尿病病人过程中如何适宜地使用胰岛素。现将结果报告如下。
1 资料和方法
1.1 一般资料
选择2016年6月—2017年8月在我院内分泌科因高尿酸血症痛风发作入院,尚未进行降尿酸或缓解疼痛治疗的病人78例(痛风组),均为男性,年龄为17~64岁,平均(41.29±13.33)岁。病人均符合2015年ACR/EULAR痛风分类标准。排除标准:骨髓增生性疾病、肿瘤放化疗后、肾脏病等所导致的继发性痛风病人。同时选择高尿酸血症无痛风并发症的门诊病人120例(高尿酸组)作为对照,均为男性,年龄17~84岁,平均(44.36±14.41)岁。两组病人均无糖尿病、严重感染及肝肾疾病,且近期未服用利尿药或影响胰岛素及尿酸代谢的药物,无大量饮酒史;两组年龄、身高等比较,差异无显著性(P>0.05)。
1.2 检测指标及方法
入院后病人禁饮食8 h后于次日的清晨采集静脉血,采用全自动生化分析仪(BECKMAN COULTER,AU5800,USA)检测血清胰岛素、尿酸、糖化血红蛋白、谷丙转氨酶/谷草转氨酶(ALT/AST)、三酰甘油、总胆固醇、高密度脂蛋白(HDL)、低密度脂蛋白(LDL)及游离脂肪酸的水平。
1.3 统计学处理
2 结 果
2.1 两组病人胰岛素等指标比较
痛风组病人空腹胰岛素水平、体质量指数、腰臀比与高尿酸组病人比较明显升高,HDL水平显著降低(t=-2.287~3.276,P<0.05);而两组病人血尿酸、糖化血红蛋白、ALT/AST、三酰甘油、总胆固醇、LDL、游离脂肪酸水平无明显统计学差异(P>0.05)。结果见表1。
表1 两组病人相关指标的比较
2.2 痛风发作相关危险因素分析
以是否发生痛风为自变量,以空腹胰岛素、体质量指数、腰臀比、HDL为协变量,将样本随机抽取90%作为训练样本,利用二元Logistic回归分析进行筛选得到两个变量与痛风发作有统计学意义。空腹胰岛素OR值大于1,表示空腹高胰岛素水平是痛风发作的危险因素;而HDLOR值小于1,表示HDL水平增高是痛风发作的保护因素。其模型参数估计见表2。
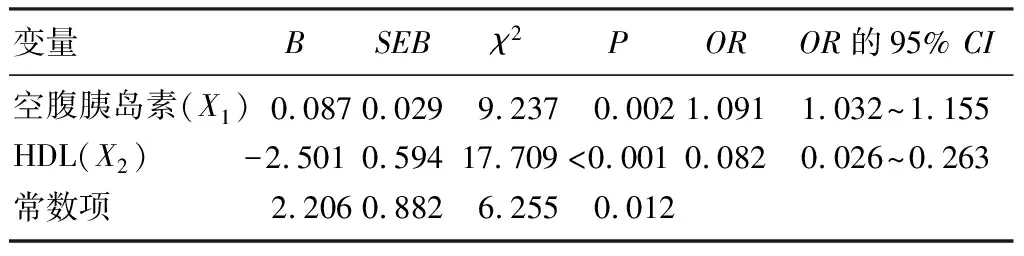
表2 Logistic回归分析结果
进而可以建立预测模型:
根据此模型可以预测痛风发作的概率Pr,然后用ROC曲线决定判断痛风发作的临界点;取临界点Pr为0.542,即当Pr≥0.542时判为痛风发作,当Pr<0.542时判为痛风不会发作;将剩余10%的样本作为测试样本回带,对预测模型进行检验,得出预测模型诊断痛风发作的灵敏度为54.5%,特异度为87.5%。
2.3 空腹胰岛素和HDL预测痛风的切点
以ROC曲线下面积0.5作为参考界值,空腹胰岛素的ROC曲线下面积为0.80(图1)。提示空腹胰岛素可用于预测痛风发作。进一步分析显示,以空腹胰岛素10.618 mU/L为最佳临界值,预测痛风发作的诊断灵敏度为80.0%,诊断特异度为65.5%,约登指数为0.455。HDL的ROC曲线下的面积则为0.381(图2),提示HDL不适用于预测痛风发作。
3 讨 论
本研究探讨了空腹胰岛素水平与痛风发作的关系。结果显示,痛风病人组的空腹胰岛素水平高于高尿酸组病人。研究表明,除了某些罕见单因素综合征外,多种危险因素直接或间接作用引起痛风,主要包括高尿酸血症、遗传学因素及药物等[2]。
肾小管功能受高胰岛素血症干扰,胰岛素介导的葡萄糖清除下降使血清尿酸清除减低,从而导致高尿酸血症[3]。血清胰岛素升高可增加肾脏近端小管对尿酸的重吸收[4];高胰岛素血症可致细胞氧化磷酸化功能受损,腺苷的浓度升高可促进尿酸的生成并增加肾脏储存尿酸的能力[5]。高水平胰岛素及胰岛素前体可促进肾小管Na+-H+交换,使尿酸重吸收相应增多而出现高尿酸血症[6]。
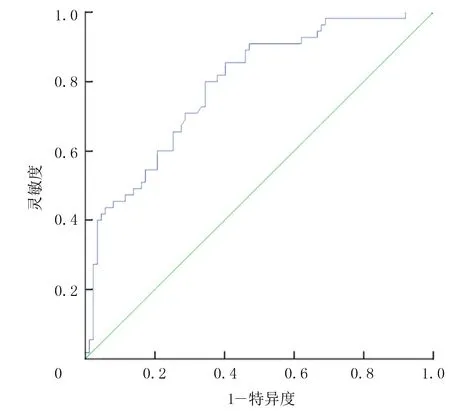
图1 空腹胰岛素的ROC曲线
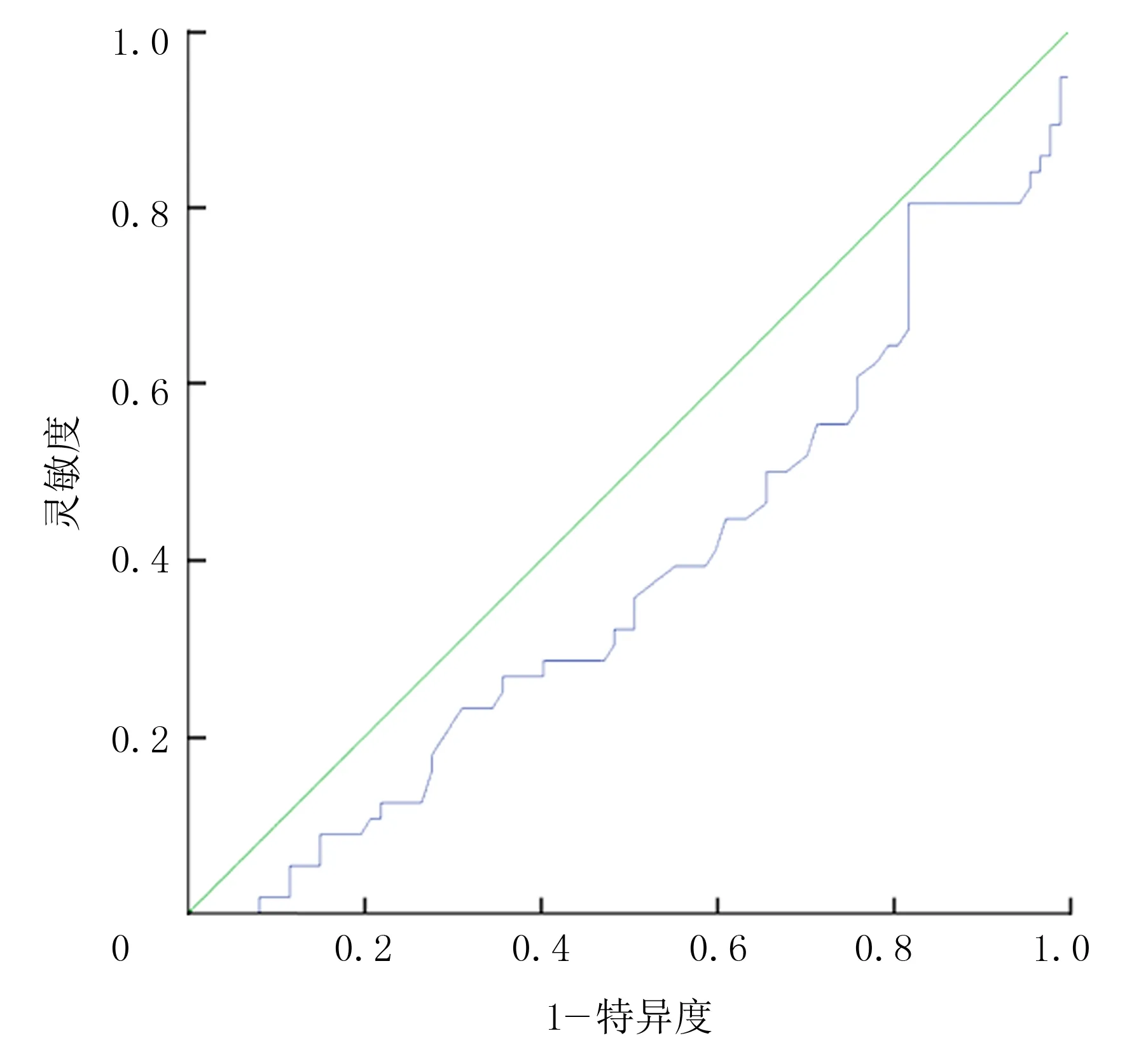
图2 HDL的ROC曲线
血清胰岛素水平升高可以激活PI3K,通过Akt/IKK信号通路激活参与非代谢器官炎症反应的核心转录因子(NF-κB)[7]。活化的 NF-κB 进入细胞核后,参与细胞炎症的过程,调节包括 TNF-α、IL-1β、IL-6、IL-8在内的炎症因子的表达[8-9],而IL-1β在痛风性关节炎发生及痛风发作的过程中发挥关键的作用[10-13]。PI3K又可以通过AKt激活mTOR,进而调节如TNF-α、IL-1β等炎症因子的分泌[14-16]。同时促炎因子又可激活JNK和IKK/NF-κB信号通路上调炎症递质水平,炎性递质水平升高又反过来激活JNK和IKK/NF-κB,两者相互影响,最终导致痛风发作[17]。高胰岛素血症还可以通过MAPK通路激活NF-κB加重炎症的反应[7],导致痛风的发作。本研究通过Logistic回归发现空腹高胰岛素水平是痛风发作的危险因素,可能是通过上述机制导致痛风的发生。本研究计算的胰岛素的ROC曲线下面积为0.800,提示空腹胰岛素可用于预测痛风的发作。
高胰岛素血症通过增加ChREBP、SREBP-1c、低度炎症反应以及TNF-α等促进肝脏合成脂质,脂质增多使新陈代谢中的核酸总量增加,加速嘌呤从头合成途径,增加尿酸生成[18]。脂肪分解为酮体的过程可阻碍尿酸排泄,使尿酸水平间接升高。关节腔内尿酸浓度过饱和形成尿酸盐结晶,从而启动痛风炎性通路,引发炎症级联反应[19]。脂质在脂肪细胞中过多的蓄积引发肥胖,从而导致脂肪组织的炎症反应[20]。激活NF-κB使脂肪细胞增加趋化因子如MCP1的合成和分泌,这导致巨噬细胞的浸润。高胰岛素血症还可以通过MAPK通路激活NF-κB加重炎症的反应[7],从而导致痛风的发作。这与本研究得出的结论一致,可能是胰岛素升高导致痛风发作的机制。
本研究还发现,HDL水平增高是痛风发作的保护因素。在促炎细胞因子介导的炎症反应中HDL也起负性调节作用。HDL水平降低,其负性调节炎症的作用也降低,可能在炎症反应中加重痛风相关的损害。
总之,本研究结果显示痛风组的空腹胰岛素水平显著高于高尿酸组,胰岛素水平是痛风发作的危险因素,胰岛素水平的升高预示着风险增加。HDL水平升高是痛风发作的保护因素。本研究进一步评估空腹胰岛素预测痛风发作的切点。结果提示,空腹胰岛素水平大于10.618 mU/L时,预测痛风发作的诊断灵敏度为80.0%,诊断特异度为65.5%。综上所述,空腹胰岛素水平有助于临床预测病人痛风发作,为预防痛风发作提供一个很好指标。
[参考文献]
[1] RICHETTE P, BARDIN T. Gout[J]. Lancet, 2010,375(9711):318-328.
[2] KUO C F, GRAINGE M J, ZHANG W, et al. Global epidemiology of gout: Prevalence, incidence and risk factors[J]. Nat Rev Rheumatol, 2015,11(11):649-662.
[3] NISKANEN L, LAAKSONEN D E, LINDSTROM J, et al. Serum uric acid as a harbinger of metabolic outcome in subjects with impaired glucose tolerance: The Finnish Diabetes Prevention Study[J]. Diabetes Care, 2006,29(3):709-711.
[4] DOSHI M, TAKIUE Y, SAITO H, et al. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice[J]. Nucleosides Nucleotides Nucleic Acids, 2011,30(12):1290-1294.
[5] SUZUKI S, YONEYAMA Y, SAWA R, et al. Relation between serum uric acid and plasma adenosine levels in women with preeclampsia[J]. Gynecol Obstet Invest, 2001,51(3):169-172.
[6] TSUNODA S, KAMIDE K, MINAMI J, et al. Decreases in serum uric acid by amelioration of insulin resistance in overweight hypertensive patients: effect of a low-energy diet and an insulin-sensitizing agent[J]. Am J Hypertens, 2002,15(8):697-701.
[7] PARK M H, KIM D H, LEE E K, et al. Age-related inflammation and insulin resistance: A review of their intricate interdependency[J]. Arch Pharm Res, 2014,37(12):1507-1514.
[8] KUO C C, LIN W T, LIANG C M, et al. Class Ⅰ and Ⅲ phosphatidylinositol 3'-Kinase play distinct roles in TLR signaling pathway[J]. J Immunol, 2006,176(10):5943-5949.
[9] SCHMIDT A M, STERN D M. Hyperinsulinemia and vascular dysfunction: the role of nuclear factor-kappaB, yet again[J]. Circ Res, 2000,87(9):722-724.
[10] MITROULIS I, KAMBAS K, RITIS K. Neutrophils, IL-1beta, and gout: Is there a link[J]. Semin Immunopathol, 2013,35(4):501-512.
[11] AMARAL F A, COSTA V V, TAVARES L D, et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout[J]. Arthritis Rheum, 2012,64(2):474-484.
[12] TORRES R, MACDONALD L, CROLL S D, et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis[J]. Ann Rheum Dis, 2009,68(10):1602-1608.
[13] CUMPELIK A, ANKLI B, ZECHER D, et al. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome[J]. Ann Rheum Dis, 2016,75(6):1236-1245.
[14] WEICHHART T, COSTANTINO G, POGLITSCH M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response[J]. Immunity, 2008,29(4):565-577.
[15] WEICHHART T, HENGSTSCHLAGER M, LINKE M. Regulation of innate immune cell function by mTOR[J]. Nat Rev Immunol, 2015,15(10):599-614.
[16] LEE P S, WILHELMSON A S, HUBNER A P, et al. mTORC1-S6K activation by endotoxin contributes to cytokine up-regulation and early lethality in animals[J]. PLoS One, 2010,5(12): e14399.
[17] PERKINS N D. Integrating cell-signalling pathways with NF-kappa B and IKK function[J]. Nat Rev Mol Cell Bio, 2007,8(1):49-62.
[18] MATSUBARA K, MATSUZAWA Y, JIAO S, et al. Relationship between hypertriglyceridemia and uric acid production in primary gout[J]. Metabolism, 1989,38(7):698-701.
[19] ZHOU Y, FANG L, JIANG L, et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway[J]. PLoS One, 2012,7(6): e39738.
[20] MCLAUGHLIN T, LIU L F, LAMENDOLA C, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans[J]. Arterioscler Thromb Vasc Biol, 2014,34(12):2637-2643.
