酸性环境抑制高磷诱导大鼠VSMCs钙化的机制研究*
2018-06-19耿同会徐金升韩迎迎白亚玲张俊霞崔立文张胜雷
耿同会,徐金升,韩迎迎,白亚玲,张俊霞,崔立文,张胜雷
(河北医科大学第四医院 肾内科,河北 石家庄 050011)
慢性肾脏病(chronic kidney disease, CKD)患者心血管疾病发病率是普通人群的10~30倍[1],血管钙化为主要危险因素[2]。血管钙化是细胞介导的生物矿化过程[3],血管平滑肌细胞(vascular smooth muscle cells, VSMCs)表型转化是其重要机制[4]。代谢性酸中毒是CKD患者常见的并发症。研究发现,酸性环境可抑制大鼠血管钙化,但具体机制尚不明确[5]。活化T细胞核因子(nuclear factor of activated T cells, NFAT)是一种有多种调控作用的转录因子,GOETTSCH等[6]发现,NFATc1对血管钙化有促进作用。因此笔者推测,酸性环境可能通过下调NFATc1表达,抑制血管钙化。本实验以大鼠VSMCs为研究对象,旨在探讨酸性环境对高磷诱导大鼠VSMCs钙化的影响及其可能机制。
1 材料与方法
1.1 材料
1.1.1 实验动物 6只4周龄健康雄性SD大鼠,原代培养大鼠胸主动脉平滑肌细胞,大鼠购自河北医科大学实验动物中心,动物合格证标号:1305090。
1.1.2 主要仪器和试剂 倒置相差显微镜(LH50A型)(日本Olympus公司),CYTATION3型酶标仪(美国Biotek公司),胎牛血清﹑细胞培养基购自美国Gibco公司,β-甘油磷酸(美国Sigma公司),酸度计(北京赛多利斯科学仪器有限公司),碱性磷酸酶(alkaline phosphatase, ALP)活性试剂盒(南京建成生物工程研究所),钙含量测定试剂盒(北京中生北控生物科技股份有限公司),RNA提取试剂盒﹑RNA逆转录试剂盒(美国Thermo公司),PCR引物(上海英潍捷基公司),NFATc1﹑Runt相关转录因子2(runt related transcription factor 2, Runx2)抗体购自英国Abcam公司,GAPDH抗体(美国Bioworld公司)。
1.2 方法
1.2.1 钙化VSMCs的制备及分组 取4周龄健康雄性SD大鼠的胸主动脉,采用组织块贴壁法培养原代VSMCs,实验取3~5代细胞。采用随机数字表法,将VSMCs随机分为正常对照组﹑高磷+pH 7.4组﹑高磷+pH 7.1组。正常对照组用含10%胎牛血清培养基培养,调整pH值至7.4;高磷+pH 7.4组在10%胎牛血清培养基中加入10 mmol/L β-甘油磷酸,调整pH值至7.4;高磷+pH 7.1组用高磷培养基,调整pH值至7.1。使用1 mol/L盐酸HCl和7.4%碳酸氢钠调整培养基pH值,换液1次/24 h。
1.2.2 细胞钙含量测定 将24孔板内培养的细胞干预14 d后,弃上清液,用1 mmol/L HCl消化过夜,取上清,使用邻甲酚酞络合酮比色法测定钙含量;细胞用0.1 mol/L氢氧化钠溶液和0.1%十二烷基硫酸钠溶解30min,根据吸光度值计算细胞蛋白含量。VSMCs的钙含量结果用细胞钙含量与蛋白含量的比值表示(mg/g)。
1.2.3 茜素红染色 取4~5代细胞以5×104个/孔密度接种于24孔板内,干预14 d。用pH 8.4浓度为0.1%的茜素红染液进行钙化染色,倒置显微镜下观察照相。结果判断标准:钙盐沉积为橘红色。
1.2.4 ALP活性测定 将细胞培养14 d后弃上清,磷酸盐缓冲溶液冲洗2次,加入1%聚乙二醇辛基苯基醚生理盐水,置于4℃冷藏24 h后离心,取上清液,按ALP活性试剂盒说明书操作,测定其活性,结果用蛋白质校准。
1.2.5 逆转录聚合酶链反应(reverse transcriptionpolymerase chain reaction, RT-PCR) RT-PCR检测VSMCs目的基因的表达。干预4 d后提取各组细胞mRNA,测定NFATc1和Runx2基因的表达。PCR引物由Primer 5.0软件设计,引物序列见表1。PCR反应体系为20μl,NFATc1反应条件:95℃预变性5min,95℃变性30 s,60℃退火30 s,72℃延伸45 s,共32个循环;GAPDH反应条件:95℃预变性5min,95℃变性30 s,58℃退火30 s,72℃延伸45 s,共32个循环;Runx2反应条件:95℃预变性5min,95℃变性30 s,55℃退火30 s,72℃延伸45 s,共28个循环,72℃继续延伸10min。实验重复3次。
1.2.6 Western blot检测 Western blot检测VSMCs目的蛋白的表达。干预4 d后提取各组细胞蛋白,测定NFATc1和Runx2蛋白的表达。细胞蛋白质用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳分离,转膜﹑封闭,加入一抗稀释液(NFATc1 1∶1 000,Runx2 1∶500,GAPDH 1∶5 000),4℃孵育过夜。洗膜,放入二抗稀释液(1∶5 000),室温下孵育1 h,定影显色,分析条带灰度值。实验重复3次。
1.3 统计学方法
数据分析采用SPSS 21.0统计软件,计量资料以均数±标准差(±s)表示,多组比较用单因素方差分析,组间两两比较用SNK-q检验;相关性分析用Pearson法,P<0.05为差异有统计学意义。
2 结果
2.1 酸性环境对高磷诱导VSMCs钙化的影响
干预14 d后,正常对照组﹑高磷+pH 7.4组﹑高磷+pH 7.1组VSMCs的钙含量分别为(47.423±10.515)﹑(119.306±13.443) 和(89.738±12.542)mg/g,经方差分析,差异有统计学意义(F=22.138,P=0.0017)。进一步两两比较经SNK-q检验,各组间比较差异有统计学意义(P<0.05)。高磷环境下大鼠VSMCs钙含量水平升高;pH值降低时VSMCs钙含量减少。
与钙含量结果类似,茜素红染色可见正常对照组VSMCs无橘红色钙盐结节出现,高磷组有大量橘红色钙盐沉积,而酸干预可使钙盐沉积减少。见图1。
2.2 酸性环境对高磷诱导VSMCs表型转化的影响
正常对照组﹑高磷+pH 7.4组﹑高磷+pH 7.1组VSMCs的ALP活性分别为(32.510±2.393)﹑(106.206±4.294)和(85.715±6.227)u/g,经方差分析,差异有统计学意义(F=40.909,P=0.000)。进一步两两比较经SNK-q检验,各组间比较差异有统计学意义(P<0.05)。高磷+pH 7.4组ALP活性高于正常对照组;酸干预后ALP活性降低。
正常对照组﹑高磷+pH 7.1组﹑高磷+pH 7.4组VSMCs的Runx2 mRNA相对表达量分别为(0.213±0.008)﹑(0.317±0.054)和(0.562±0.070),经方差分析,差异有统计学意义(F=36.924,P=0.000)。正常对照组﹑高磷+pH 7.1组﹑高磷+pH 7.4组VSMCs的Runx2 蛋白相对表达量分别为(0.390±0.108)﹑(0.785±0.069)和(1.095±0.121),经方差分析,差异有统计学意义(F=36.408,P=0.000)。进一步两两比较经SNK-q检验,正常对照组﹑高磷+pH 7.1组Runx2 mRNA和蛋白表达水平较高磷+pH 7.4组低(P<0.05),表明酸性环境可下调Runx2表达。见图2。
2.3 酸性环境对高磷诱导的VSMCs中NFATc1表达的影响
正常对照组﹑高磷+pH 7.1组﹑高磷+pH 7.4组VSMCs的NFATc1 mRNA相对表达量分别为(0.264±0.043)﹑(0.419±0.073 )和(0.668±0.097),经方差分析,差异有统计学意义(F=22.512,P=0.002)。正常对照组﹑高磷+pH 7.1组﹑高磷+pH 7.4组VSMCs的NFATc1蛋白相对表达量分别为(0.155±0.066)﹑(0.362±0.031)和(0.661±0.096),经方差分析,差异有统计学意义(F=36.408,P=0.000)。进一步两两比较经SNK-q检验,正常对照组﹑高磷+pH 7.1组NFATc1 mRNA和蛋白表达水平较高磷+pH 7.4组低(P<0.05),高磷环境NFATc1表达升高。见图3。
2.4 NFATc1与Runx2、ALP的相关性
VSMCs中NFATc1与Runx2表达呈正相关(r=0.801,P=0.003),NFATc1与ALP活性呈正相关(r=0.698,P=0.002)。

图1 各组大鼠VSMCs钙盐沉积 (茜素红染色)
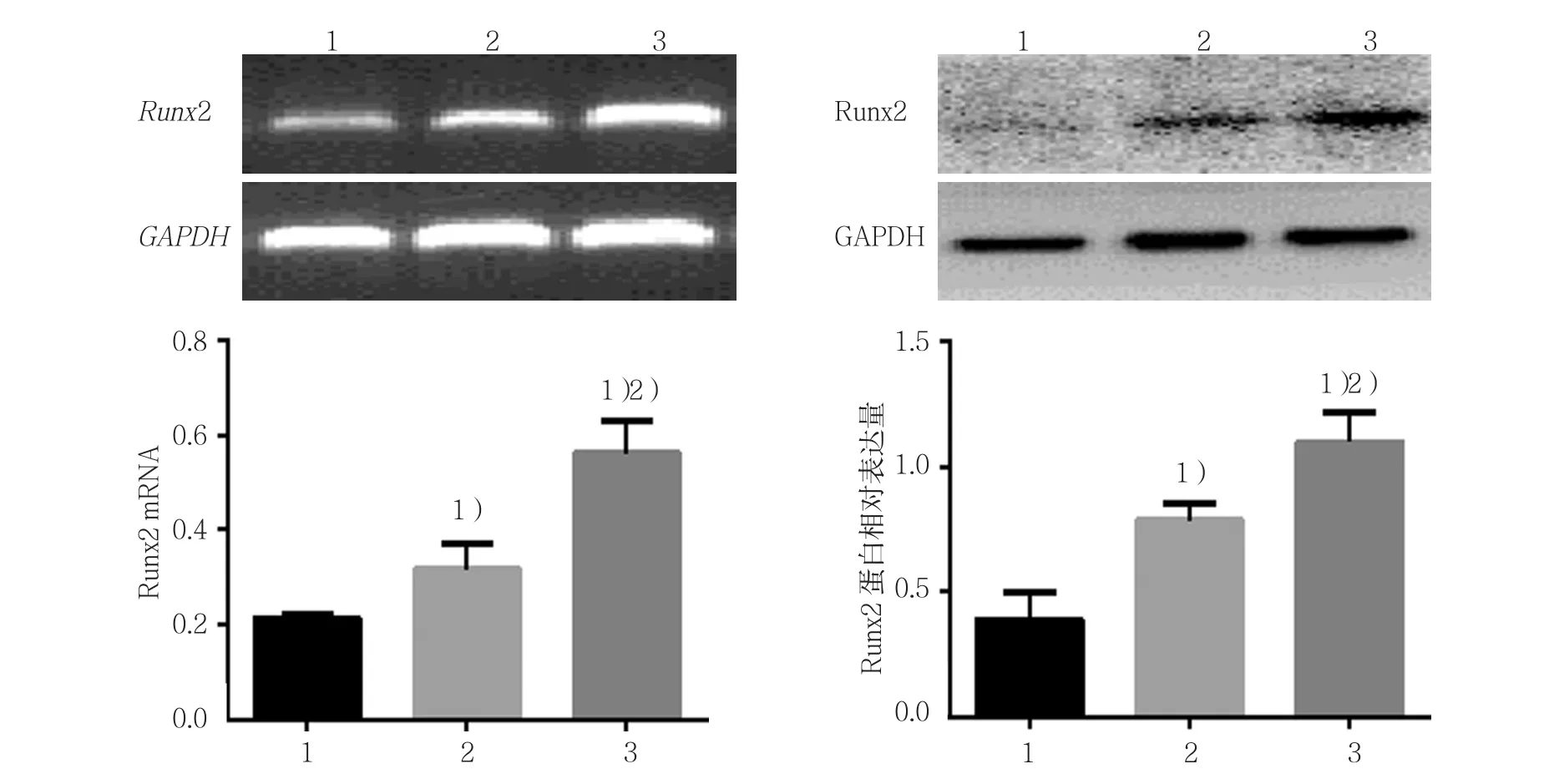
图2 各组大鼠VSMCs的Runx2 mRNA和蛋白的表达

图3 各组大鼠VSMCs的NFATc1 mRNA和蛋白的表达
3 讨论
代谢性酸中毒是CKD患者的常见并发症。近期研究发现,代谢性酸中毒可以抑制VSMCs表型转化[4]。近年来就酸性环境对骨形成的作用研究较为深入,然而其对血管钙化的作用研究甚少,仅证实VSMCs成骨/成软骨表型转化是其重要机制之一[7],因此,可将酸性环境对成骨作用的影响推演到血管钙化上。血管钙化是CKD患者发生心血管疾病的重要危险因素,不同于普通人群发生的动脉粥样硬化,CKD患者以血管中膜钙化为突出表现[8]。VSMCs作为血管中膜的重要成分,在血管钙化发生过程中起重要作用。本实验通过体外培养VSMCs,深入探讨酸性环境对VSMCs钙化的影响,发现与正常对照组相比,高磷刺激加重VSMCs钙化,酸刺激可抑制高磷诱导的VSMCs钙化。
血管钙化是多因素共同参与的复杂病理过程,其发病机制尚未完全清楚。血管钙化常常伴随Runx2和ALP等成骨相关蛋白的表达[7]。Runx2是Runt结构域基因家族一员,可结合成骨细胞特异性顺式作用元件,调控下游骨基质和骨胶原的产生,参与成骨细胞分化[7]。ALP亦是成骨细胞形成的早期标志物。两者在正常VSMCs中表达较低,在钙化的血管和心脏瓣膜组织中表达升高[9]。因此,可以凭借VSMCs中Runx2的表达量和ALP活性推测其是否发生成骨样表型转化。本研究结果显示,与高磷+pH 7.4组相比,高磷+pH 7.1组VSMCs的Runx2表达和ALP活性降低,提示高酸性环境抑制VSMCs钙化的机制可能是抑制VSMCs发生成骨样表型转化。
NFAT最初在T细胞中因其免疫调控作用被发现,之后发现除免疫系统外,NFAT转录因子在其他组织也有广泛分布,如心肌﹑骨骼肌﹑卵巢等组织,具有多种生理功能,如参与刺激T细胞活化﹑成骨细胞分化和成骨作用﹑破骨细胞分化,调控内皮细胞及VSMCs增殖[10]。NFAT家族蛋白活性主要由Ca2+/钙调神经磷酸酶(Calcineurin, CaN)调节。当Ca2+﹑钙调节蛋白与CaN结合后诱发CaN构象改变,使自身抑制域移位,暴露出活性位点,导致CaN活化,继而使细胞浆中NFAT脱磷酸化,暴露其核定位信号,NFAT得以转位入核,与核内其他转录因子协同调节多种基因的活化[11]。研究表明,在VSMCs中,NFATcl高表达,NFATc3少量表达,而NFATc2和NFATc4几乎不表达[12]。有研究发现,在氧化低密度脂蛋白诱导VSMC钙化过程中,NFAT发挥重要作用,敲除NFAT基因能抑制Runx2表达和基质矿化[6]。以上研究表明,NFATc1在血管钙化中发挥重要作用,因此笔者推测NFATc1可能参与酸抑制VSMCs钙化的过程。本研究从体外实验观察VSMCs中NFATc1的表达,结果发现高磷+pH 7.4组VSMCs中NFATc1表达高于正常对照组,提示NFATc1参与慢性肾衰竭大鼠血管钙化的发生,可能对其有促进作用;进一步比较发现,酸性环境能够抑制VSMCs中NFATc1 mRNA和蛋白的表达,由此推测NFATc1可能是酸抑制VSMCs钙化发生的因素之一。
NFATc1在酸抑制高磷诱导的VSMCs钙化中如何发挥作用?本研究就VSMCs中NFATc1蛋白表达水平与表型转化指标(ALP活性和Runx2蛋白表达水平)做相关性分析,结果发现NFATc1与Runx2蛋白表达﹑ALP活性呈正相关。因此,笔者推测酸抑制VSMCs发生钙化的可能机制是通过降低NFATc1表达,抑制VSMCs发生表型转化来实现的。
综上所述,本研究通过体外研究发现,酸性环境可以抑制高磷诱导的大鼠VSMCs钙化,其可能机制是通过降低NFATc1表达,抑制VSMCs发生表型转化来实现的。但本研究仅从实验室内观察到NFATc1在酸抑制血管钙化中的作用,具体机制仍需进一步实验研究,以期为临床预防和治疗CKD患者的血管钙化提供新靶点。
参 考 文 献:
[1]JHA V, GARCIA-GARCIA G, ISEKI K, et al. Chronic kidney disease: global dimension and perspectives[J]. Lancet, 2013,382(9888): 260-272.
[2]白亚玲, 徐金升, 武娇, 等. 慢性肾脏病大鼠血管收缩张力与血管钙化的关系[J]. 中国现代医学杂志, 2014, 24(11): 19-23.
[3]LIU Y, LIN F, FU Y, et al. Cortistatin inhibits calcification of vascular smooth muscle cells by depressing osteoblastic differentiation and endoplasmic reticulum stress[J]. Amino Acids,2016, 48(11): 2671-2681.
[4]管思明, 辛华平, 方欣, 等. 血管中骨髓间充质干细胞在不同钙化环境中的分化研究[J]. 中华老年医学杂志, 2014, 33: 916-919.
[5]MENDOZA F J, LOPEZ I, MONTES de OCA A, et al. Metabolic acidosis inhibits soft tissue calcification in uremic rats[J]. Kidney Int, 2008, 73(4): 407-414.
[6]GOETTSCH C, RAUNER M, HAMANN C, et al. Nuclear factor of activated T cells mediates oxidised LDL-induced calcification of vascular smooth muscle cells[J]. Diabetologia, 2011, 54: 2690-2701.
[7]DEMER L L, TINTUT Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification[J]. Arterioscler Thromb Vasc Biol, 2014, 34: 715-723.
[8]LANZER P, BOEHM M, SORRIBAS V, et al. Medial vascular calcification revisited: review and perspectives[J]. Eur Heart J,2014, 35: 1515-1525.
[9]ZHOU S, FANG X, XIN H, et al. Osteoprotegerin inhibits calcification of vascular smooth muscle cell via down regulation of the Notch1-RBP-Jkappa/Msx2 signaling pathway[J]. PLoS One,2013, 8(7): DOI: 10.1371/journal.pone.68987.
[10]QIN J J, NAG S, WANG W, et al. NFAT as cancer target: mission possible[J]. Biochim Biophys Acta, 2014, 1846: 297-311.
[11]CHOO M K, YEO H, ZAYZAFOON M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation[J]. Bone, 2009, 45:579-589.
[12]WADA H, HASEGAWA K, MORIMOTO T, et al. Calcineurin-GATA-6 pathway is involved in smooth muscle-specific transcription[J]. J Cell Biol, 2002, 156: 983-991.
