高效液相色谱波长切换法同时测定百效丸中6种主成分的含量
2018-06-07张铭马丽颖
张铭,马丽颖
(辽宁中医药大学附属医院制剂中心,辽宁 沈阳 110032)
百效丸由玄参、巴豆霜、厚朴、川贝母、柴胡等12味中药材加工而成,为棕红色的大蜜丸,气香、味凉、微辛,具有消积理滞、镇惊化痰的功效,临床用于寒热凝结、停食宿水、腹疼腹胀、痰喘气促、慢惊风症等。百效丸收载于《中药成方制剂第二册》,现行质量标准未对百效丸中任何成分进行定量测定[1],也未检索到对该制剂进行多组分测定的文献报道,为全面控制本品质量,本研究采用高效液相色谱(HPLC)波长切换法同时测定处方中玄参[2]主成分哈巴苷、安格洛苷C和哈巴俄苷,巴豆霜主成分巴豆苷,厚朴[2]主成分和厚朴酚[3]、厚朴酚的含量,为提高百效丸质量标准提供了数据支持。
1 仪器与试药
1.1仪器高效液相色谱仪(型号:Agilent 1100系列,生产厂家:美国安捷伦公司); 电子天平(型号:AXS205DU型,生产厂家:梅特勒-托利多仪器有限公司);数控超声波清洗器(型号:KQ3200DB型,生产厂家:昆山市超声仪器有限公司)。
1.2试药与试剂百效丸来源于天津达仁堂京万红药业有限公司,批号分别为162156、162175、162182;乙腈、甲醇为色谱纯,其它试剂为分析纯;哈巴苷对照品(111729-201506)、哈巴俄苷对照品(111730-201508)、巴豆苷对照品(111856-201001)、和厚朴酚对照品(110730-201614)、厚朴酚对照品(110729-201513)均购于中国食品药品检定研究院;安格洛苷C对照品(115909-22-3)购于上海宝曼生物科技有限公司。
2 方法与结果
2.1色谱条件采用Venusil MP C18(4.6 mm×250 mm,5 μm)色谱柱;流动相A:乙腈-甲醇(1∶4),流动相B:0.2%磷酸溶液,梯度洗脱(0~20 min,48.0%A;20~26 min,48.0%A→54.0%A;26~35 min,54.0%A→60.0%A;35~49 min,60.0%A→75.0%A;49~55 min,75.0%A→48.0%A);0~20 min时在210 nm[4-7]波长下检测哈巴苷,20~26 min在280 nm[4,6,8-9]波长下检测安格洛苷C和哈巴俄苷;26~55 min在294 nm[10-11]波长下检测巴豆苷、和厚朴酚、厚朴酚;柱温:30 ℃;进样量为10 μL;流速:0.9 mL·min-1。
2.2溶液的制备
2.2.1对照品溶液 分别精密称取对照品哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚各适量,用50%甲醇分别制成每毫升中含哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚各为0.796、0.530、0.490、1.382、1.058、1.874 mg的单成分对照品储备溶液。再分别依次量取2.5、1.0、1.0、2.5、2.5、2.5 mL,置同一量瓶中(50 mL),用50%甲醇稀释制成百效丸检测用混合对照品溶液。
2.2.2百效丸供试品溶液 取百效丸适量,剪碎,称取4.0 g,精密称定,将其置于具塞锥形瓶中,精密加入50%甲醇50 mL,称定其重量,KQ3200DB型数控超声波清洗器超声提取30 min,放冷,用50%甲醇补足减失重量,摇匀,滤过,取续滤液,即得百效丸供试品溶液。
2.2.3阴性样品溶液 按百效丸的处方和工艺过程,分别制备不含玄参、巴豆霜、厚朴(姜制)的阴性样品,再按“2.2.2” 项下的方法制成玄参阴性样品溶液、巴豆霜阴性样品溶液、厚朴阴性样品溶液。
2.3专属性试验精密吸取“2.2.1~2.2.3”项下的混合对照品溶液、百效丸供试品溶液、玄参阴性样品溶液、巴豆霜阴性样品溶液和厚朴阴性样品溶液各适量,按上述方法进行检测。结果阴性样品色谱图中,在对照品混合液所显示的哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚对应的位置无干扰,各峰之间的分离度大于1.5,理论塔板数均大于3 500,色谱图详见图1。


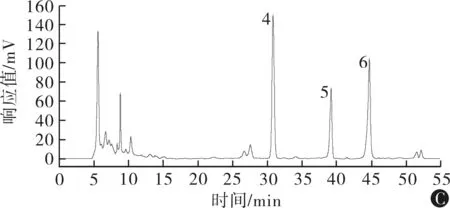

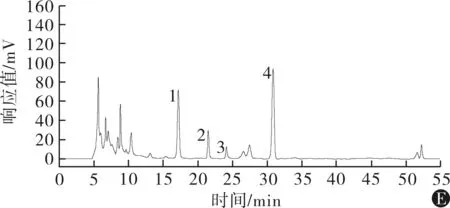
注:1为哈巴苷,2为安格洛苷C,3为哈巴俄苷,4为巴豆苷,5为和厚朴酚,6为厚朴酚
2.4线性关系考察分别精密吸取“2.2.1”项下对照品储备液各0.1、0.2、0.5、1.0、1.5、2.0 mL,分别用50%甲醇稀释至20 mL,振摇均匀,制成20倍浓度差的6种不同浓度的混合对照品溶液,依法测定,以哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚及厚朴酚的质量浓度X为横坐标,测得的哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚峰面积Y为纵坐标,绘制标准曲线,得回归方程,如表1所示。

表1 线性关系考察结果
2.5加样回收率试验分别精密称取对照品哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚及厚朴酚各适量,用50%甲醇分别溶解并稀释制成含哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚及厚朴酚为0.916、0.323、0.281、0.807、0.719、0.596 g·mL-1单一成分对照品溶液,备用。
取百效丸(批号162156)6份,剪碎,每份约2.0 g,精密称定,置具塞锥形瓶中,依次精密加入上述备用对照品溶液1.0、1.0、1.0、2.0、2.0、4.0 mL,再按“2.2.2”的方法制备加样回收样品溶液,依法测定,计算哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚及厚朴酚的回收率及相应的RSD,结果它们的回收率与相应的RSD见表2~4。
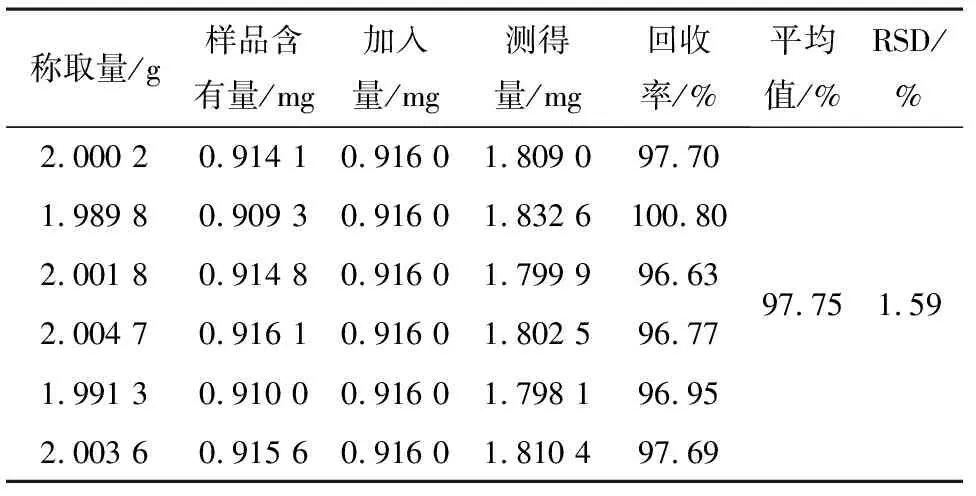
表2 哈巴苷加样回收率实验结果

表3 安格洛苷C加样回收率实验结果

表4 哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚加样回收率实验结果
2.6重复性试验取百效丸样品(批号162156),按“2.2.2”项下制备方法制备百效丸供试品溶液6份,按“2.1”项下的色谱条件及检测方法进行测定,分别计算哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚的含量,结果所测6种组分含量的RSD依次为0.42%、1.51%、0.76%、1.13%、1.24%和0.90%。
2.7精密度试验取“ 2.2.1”项下的混合对照品溶液,连续进样6次,依法测定各成分峰面积,结果哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚峰面积的RSD (n=6)分别为1.16%、0.25%、1.29%、0.93%、1.10%和0.82%。
2.8供试品溶液稳定性试验取同一份百效丸供试品溶液,于室温下0 h、2 h、4 h、6 h、8 h、10 h进样测定哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚的峰面积值,结果室温下10 h内稳定,所测6种组分峰面积的RSD分别为1.18%、0.74%、1.30%、0.96%、1.05%和0.54%。
2.9样品测定取三批百效丸样品,按“2.2.2”项下制备方法制备百效丸供试品溶液,依法进样测定结果见表5。
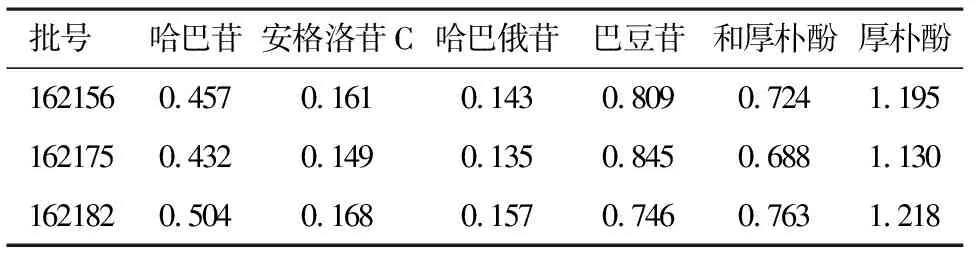
表5 含量测定结果/mg·g-1
3 讨论
3.1流动相的选择在试验过程中,作者首先考察了甲醇-水[12-13]、乙腈-水[4]、乙腈-甲醇(1∶4)与水[14-16]流动相体系,以色谱峰基线平稳情况和所测各成分的峰形、分离效果为指标,优选最佳的流动相体系。结果显示乙腈-甲醇(1∶4)与水流动相体系优于甲醇-水流动相体系和乙腈-水流动相体系,但个别色谱峰峰形和分离效果还欠佳。在此基础上,作者又考察了乙腈-甲醇(1∶4)与1.0%冰醋酸溶液[5]、乙腈-甲醇(1∶4)与2.0%冰醋酸溶液[11]、乙腈-甲醇(1∶4)与0.2%磷酸溶液[17]、乙腈-甲醇(1∶4)与0.5%磷酸溶液[10]流动相体系,实验结果显示以乙腈-甲醇(1∶4)与0.2%磷酸溶液作为流动相按照文中的梯度洗脱比例进行洗脱时,所测各成分哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚峰形较好,各色谱峰之间以及各色谱峰与杂质峰之间均能达到有效分离。
3.2提取方法的选择在提取方式的选择上,作者分别尝试了加热回流提取和超声提取,结果加热回流提取和超声提取效果差异不大,考虑到检验过程操作的便捷性,优选超声提取作为百效丸供试品溶液的提取方式;在此基础上,作者又以所测成分哈巴苷、安格洛苷C、哈巴俄苷、巴豆苷、和厚朴酚、厚朴酚提取率为指标,对比考察了超声提取时间(15、30、45 min)和提取溶剂(水、50%甲醇、70%甲醇)。实验结果表明,以50%甲醇为提取溶剂时,所测各成分综合提取率最佳;超声提取15 min时,部分所测成分含量偏低,超声提取30 min和超声提取45 min所测各成分含量差异无统计学意义。故本文采用50%甲醇超声提取30 min作为百效丸供试品溶液的制备方法。
4 小结
本研究建立的百效丸中6种主成分含量同时测定的方法,填补了本品在液相色谱定量分析方面的空白,可进一步保证百效丸产品质量和临床疗效,所建立的方法操作简便,重复性好。但也存在着一定的不足之处,实验过程中对照品使用量较大,检验成本偏高,采用一测多评法同时测定百效丸中多成分含量还在进一步研究中。
[1] 国家药典委员会.卫生部颁药品标准(中药成方制剂第二册)[S].北京:人民卫生出版社,1990:94.
[2] 国家药典委员会.中国药典(2015年版一部)[S].北京:中国医药科技出版社,2015:81,117,251-252.
[3] 王冠杰,赵娅娟,董济民.脂质体和厚朴酚联合热疗对腺样囊性癌增殖的协同抑制作用[J].安徽医药,2016,20(7):1237-1240.
[4] 刘利利,刘颖新,毛群芳,等.HPLC法同时测定苍术-玄参药对中4种活性成分的含量[J].药物分析杂志,2016,36(1):81-85.
[5] 周小雅,赖飞娥,刘雪梅.高效液相色谱-紫外双波长法同时测定玄参配方颗粒中3种主要成分的含量[J].中国中医药信息杂志,2014,21(5):78-80.
[6] 程晓莉.HPLC测定中药材玄参中有效成分的含量分析[J].实用中医内科杂志,2012,26(11):6-7.
[7] 王海丽,蒙蒙.HPLC法测定儿童回春颗粒中哈巴苷和哈巴俄苷的含量[J].西北药学杂志,2016,31(6):584-587.
[8] 舒柯.双波长HPLC法同时测定玄麦甘桔颗粒中哈巴苷和哈巴俄苷的含量[J].中国民族民间医药,2016,25(10):31-32,36.
[9] 钱永健,张超.HPLC法测定出血热预防片中的女贞苷、特女贞苷、哈巴苷和哈巴俄苷[J].中成药,2015,37(1):116-120.
[10] 马鹏,林涛.反相高效液相色谱法测定和比较厚朴及和厚朴中厚朴酚及和厚朴酚含量[J].华西药学杂志,1993,8(8):87-89.
[11] 冯家龙.HPLC法测定十香止痛丸中厚朴酚与和厚朴酚的含量[J].中国药师,2012,15(10):1503-1504.
[12] 郭建华,刘佳乐,黄亮,等.高效液相色谱法同时测定厚朴提取物中厚朴酚与和厚朴酚[J].广东化工,2014,17(41):181-182.
[13] 赵呈雷,黄一平,田耀洲,等.HPLC法同时测定桂朴微丸中厚朴酚、和厚朴酚的含量[J].中国药房,2016,27(33):4695-4697.
[14] 凌云,赵红玉,潘兴亮,等.高效液相色谱法测定巴豆中巴豆苷的含量[J].中西医结合学报,2009,7(11):1057-1060.
[15] 陈彦琳,金峰,杜杰,等.不同制霜方法制备巴豆霜饮片质量比较[J].中国现代中药,2015,17(11):1201-1203,1232.
[16] 邱远金,朱国强,贾晓光,等.HPLC法测定抗结核病药物四味抗痨丸中巴豆苷的含量[J].西北药学杂志,2015,30(1):28-30.
[17] 胡静,王艳,宋丽丽,等.HPLC测定中药巴豆中巴豆苷和木兰花碱的含量[J].中国实验方剂学杂志,2015,21(8):39-42.
