核壳结构的Co3O4@δ-MnO2/Pt作为锂氧电池高效催化正极
2018-06-06曹高劭赵新兵
成 浩 谢 健*,, 陈 振 涂 健 曹高劭 赵新兵,
(1浙江大学材料科学与工程学院,杭州 310027)(2浙江省电池新材料与应用技术研究重点实验室,杭州 310027)(3湖南立方新能源科技有限责任公司,株洲 412000)
Lithium-oxygen(Li-O2)batteries now have captured a world-wide attention due to the extremely high theoretical energy density of 3 505 Wh·kg-1,showing promising applications in electric vehicles[1-10].In nonaqueous Li-O2batteries,during discharge,oxygen is reduced to O2-which reacts with Li+to form Li2O2at cathode.Upon the subsequent recharge,Li2O2can be electrochemically decomposed to release oxygen and Li+[4].Unlike the shuttle mechanism in Li-ion batteries,the deposition of insulating/insoluble Li2O2will cause sluggish oxygen reduction/evolution reaction(ORR/OER)kinetics,leading to high polarization,low yieldable capacity and poor cycling stability[11-12].It is widely acknowledged that the electrode kinetics can be enhanced by using efficient catalysts.Besides the components,the architecture of catalytic electrode should also be specially designed to adapt the Li2O2deposition without drastically changing the structure of the electrodes[13-17].
Carbon materials have been widely used as the catalysts for Li-O2batteries due to the low cost,high electronic conductivity,light weight and easy manipulation of the porosity[18-23].However,carbon materials suffer from decomposition chemically or electrochemically in contact with Li2O2or LiO2[24-25].In addition,the catalytic performance of carbon materials for OER is not satisfactory.Although noble metals provide the best catalytic activity for both ORR and OER[26-33],the high cost limits their large-scale applications.Transition metals oxides,such as MnO2,Co3O4,NiO,and CoOOH,are suitable due to low cost,structural stability and relatively high catalytic activity for ORR/OER[34-42].Thus,a compromise can be made by combining transition metals oxides and noble metals.However,unlike carbon materials,it is difficult to allocate free space for Li2O2deposition in oxides.
Array-type electrode prepared by direct growth route is desirable since the voids between the arrays could be used to storeCui et al.[37]first report a Co3O4-array-catalyzed Li-O2cell with low polarization and high capacity.The work by Chang et al.[43]show that Li-O2cell could sustain stable cycling up to 300 times at 500 mAh·g-1when using a carbon/binder-free RuOx/TiN nanotube arrays cathode.Recent report by Liu et al.[44]show that Li-O2batteries with TiO2-array cathode demonstrate long cycle life,superior rate capability,high round-trip efficiency,and good recoverability of the catalytic electrode.For the array-type electrode,realizing conformal growth of Li2O2on the arrays is desirable to retain the voids between the arrays and keep intimate contact between Li2O2and catalyst.Nevertheless,as previously reported,conformalgrowth ofinsulating Li2O2willeasily deactivate the catalyst with low capacity due to blocked electron transport[47-50].
In this work,we design a unique core-shell Co3O4@δ-MnO2/Pt arrays-type electrode on nickel foam by a controllable,facile route as binder-free electrodes for Li-O2cells,where the Co3O4acts as the “core” and ultrathin δ-MnO2nanosheets with Pt as the “shell”.MnO2was selected because of its high catalytic activity for ORR/OER[34-36,51].Co3O4acts both as the catalytically active component and as the substrate for MnO2deposition.The decoration of Pt not only enhances the catalytic activity butalso guides conformal growth of Li2O2.The advantages of this electrode design include:(1)the array-type structure facilitates the electrode wetting by electrolyte and O2transport and supplies large room for Li2O2loading;(2)the conformalgrowth of thin-layered Li2O2on Co3O4@δ-MnO2/Pt enables its easy decomposition upon charge;(3)the porous structure of δ-MnO2assures high Li2O2loading despite the conformal growth mode;(4)the side reactions related to binder and conductive agent are totally precluded or largely reduced due to binder-and conductive-agent-free electrode configuration.As a result,Li-O2cells with core-shell Co3O4@δ-MnO2/Pt arrays exhibit high capacity and long cycle life.
1 Experimental
1.1 Electrode preparation
Co3O4@δ-MnO2/Ptarrays-type electrodes were synthesized by three steps.First,Co3O4nanowire arrays on Ni foam substrate were prepared by a facile hydrothermal route.Co(NO3)2·6H2O(1.2 mmol),NH4F(1.2 mmol),and urea(3 mmol)were dissolved into 50 mL of deionized (DI)water under vigorous stirring.The solution was then transferred into a Teflon-lined stainless steel autoclave with a piece of Ni foam immersed.The autoclave was sealed and heated in an electric oven at 120℃for 5 h.After being cooled to room temperature,the Ni foam piece with a pink deposit was collected,washed with DI water and absolute ethanol several times,and dried at 60℃in air overnight.The product was further heated at 400℃for 2 h in air to obtain Ni-supported Co3O4(Co3O4/Ni).For the growth of δ-MnO2in the second step,a piece of Co3O4/Ni was first immersed into a 0.04 mol·L-1aqueous solution of glucose for 24 h,followed by carbonization at 450℃in Ar for 2 h.After that,80 mg KMnO4(99.5%,Sinopharm Chemical Reagent Co.,Ltd.)was dissolved in 60 mL of DI water with vigorous stirring to form a homogeneous solution.The carbon modified Co3O4/Ni was soaked in the above solution for 1.5 h.Afterwards,the mixture was transferred into a Teflon-lined stainless steel autoclave and heated at 85℃for 2.5 h.After cooling down to room temperature naturally,the Ni foam with a brown deposit was collected and washed repeatedly with DI water and absolute ethanol.The electrode was then dried at 60℃in air overnight followed by heating at 300℃in Ar for 2 h to obtain Ni-supported core-shell Co3O4/δ-MnO2.Finally,for the platinum deposition,H2PtCl6·6H2O was dispersed in DI water at a concentration of 0.24 mg·mL-1under stirring,a piece of Ni-supported Co3O4@δ-MnO2was soaked in the above solution overnight.The electrode was then dried at 60℃in air for 5 h followed by heating at 300℃in Ar for 2 h to get Ni-supported Co3O4@δ-MnO2/Pt.The total loading of Co3O4@δ-MnO2/Pt on Ni substrate is around 1.4 mg·cm-2and Pt loading is around 0.2 mg·cm-2.
1.2 Electrode characterization
X-ray diffraction(XRD)patterns of the electrodes were acquired using a Rigaku D/Max-2550pc powder diffractometer equipped with Cu Kα radiation(λ=0.154 1 nm).The operating voltage and current were 40 kV and 250 mA,respectively,and 2θ=10°~80°.The morphologies of the pristine and cycled electrodes were observed by field-emission scanning electron microscopy (SEM)usingan S-4800 microscope(Hitachi,Japan)at an accelerating voltage of 5 kV.Transmission electron microscopy (TEM)and highresolution TEM (HRTEM)were conducted on a JEM 2100F microscope at an accelerating voltage of 200 kV.X-ray photoelectron spectra (XPS)ofthe discharged and charged electrodes were collected on a KRATOS AXIS ULTRA-DLD spectrometer with Al Kα radiation(hν=1 486.6 eV).To analyze the cycled electrodes by SEM,TEM and XPS,the electrodes or electrode components were carefully handled before the various ex-situ characterizations[52].
1.3 Electrochemical measurements
Coin-type Li-O2cells were assembled in the Arfilled glovebox using lithium foil as the anode,Nisupported Co3O4@δ-MnO2/Ptasthe cathode,and Celgard C480 membrane as the separator.The electrolyte was 1 mol·L-1LiClO4(≥99.99%,Sigma-Aldrich)in tetraethylene glycol dimethyl ether(TEGDME).The cathodes were dried at 80℃in vacuum overnight before cell assembly.The assembled cells were purged with pure O2for 20 min and stayed at open voltage circuit(OCV)for 5 h before the electrochemical tests.Charge and discharge cycling was performed on a Neware battery cycler(Shenzhen,China)in a voltage window of 2.0~4.5 V(vs Li/Li+).The specific capacity(mAh·g-1)and current density (mA·g-1)of the cells were calculated based on the weight of Co3O4@δ-MnO2/Pt.For the cells using Co3O4and Co3O4@δ-MnO2catalysts,the specific capacity and current density were calculated based on the weight of Co3O4and Co3O4@δ-MnO2,respectively.Electrochemical impedance spectroscopy(EIS)measurements were conducted on the VersaSTAT3 electrochemistry workstation by applying an AC signal of 5 mV amplitude over the frequency range 10-2to 105Hz.All of the electrochemical tests were performed at 25℃.
2 Results and discussion
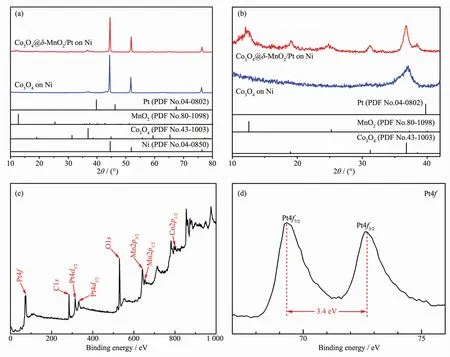
Fig.1 (a)XRD patterns of Co3O4and Co3O4@δ-MnO2/Pt on Ni foam;(b)Enlarged view in(a);(c)XPS survey spectrum and(d)Pt4f XPS spectrum of Co3O4@δ-MnO2/Pt
Fig.1a and the enlarged view (Fig.1b)show the XRD patterns of Co3O4and Co3O4@δ-MnO2/Ptsupported on Ni foam.The three strong diffraction peaks at 44.5°,51.8°,and 76.4°(2θ)are from the nickel substrate,and the presence of Co3O4and Co3O4@δ-MnO2/Pt is confirmed by XRD patterns in Fig.1b,while the presence of Pt is not detected in XRD due to the low content.The XRD shows that the MnO2is δ-MnO2.In order to further confirm the existence of Pt,XPS analysis was performed.The survey spectrum in Fig.1c reveals the presence of the expected O,Co,Mn and Pt elements.In Fig.1d,the bands at 69.3 and 72.7 eV correspond to the binding energy of Pt4f7/2and Pt4f5/2,respectively[53].In Fig.S1a,the peaks at 778.6 and 794.1 eV are the Co2p spectra of Co3+,and the peaks at 780.3 and 796.0 eV are Co2p of Co2+[54].The bands at 639.8 and 651.4 eV (Fig.S1b)correspond to the binding energy of Mn2p3/2and Mn2p1/2,in good accordance with the previous report[55].
The morphology and structural features of the prepared electrodes were characterized by SEM and TEM.Fig.2 shows the SEM images of Co3O4,Co3O4@δ-MnO2,and Co3O4@δ-MnO2/Pt at different magnifications.Note that Co3O4nanowires grow uniformly on the skeletons of the Ni foam.The Co3O4nanowires have a diameter below 200 nm and a length of several microns(Fig.2(a,b)).After the MnO2growth,thin MnO2nanosheets are uniformly covered on the whole surface of the Co3O4nanowires,forming a core-shell porous structure as shown in Fig.2(c,d).As seen in Fig.2(e,f),the introduction of Pt does not change the morphology of core-shell Co3O4@δ-MnO2obviously with the array structure maintained.The pores in Pt/δ-MnO2and the voids between the arrays are beneficial to the electrolyte infiltration and oxygen gas transportation and provide the space for Li2O2deposition.The Co3O4@δ-MnO2/Pt was further characterized by TEM,HRTEM and energy dispersive X-ray spectroscopy (EDS)mapping as shown in Fig.3.The results confirm that Co3O4,δ-MnO2and Pt construct a uniform electrode,where Co3O4exhibits a polycrystalline nanowire structure,δ-MnO2has a sheet-like shape and Pd nanocrystals have a small size below 20 nm.In Fig.3f,the lattice spacings of 0.46,0.22 and 0.21 nm correspond to the planes of Co3O4(111),Pt(111)and δ-MnO2(112),respectively.The above characterizations confirm the formation of Co3O4@δ-MnO2/Pt.

Fig.2 SEM images of(a,b)Co3O4,(c,d)Co3O4@δ-MnO2,and(e,f)Co3O4@δ-MnO2/Pt

Fig.3 (a)Dark-field TEM image,(b~e)EDS mappings in the selected area and(f)HRTEM images of Co3O4@δ-MnO2/Pt
The electrocatalytic activity of Co3O4@δ-MnO2/Pt was investigated in Li-O2cells and compared with that of Co3O4and Co3O4@δ-MnO2.The current density and specific capacity were calculated by the total mass of catalyst on the Ni substrate.The cycling performance of the Co3O4,Co3O4@δ-MnO2and Co3O4@δ-MnO2/Ptcatalyzed Li-O2cells was evaluated by galvanostatic cycling at 200 mA·g-1between 2.0~4.5 V with a limited capacity of 500 mAh·g-1.As shown in Fig.4(a,b),the cell with Co3O4@δ-MnO2/Pt catalyst can sustain stable cycling for 65 cycles.While for the cells with Co3O4and Co3O4@δ-MnO2catalytic cathodes,the cycling can last only 10 cycles and 28 cycles,respectively(Fig.4a and Fig.S2).To demonstrate the high catalytic activity of Co3O4@δ-MnO2/Pt,the cell was also cycled at a higher limited capacity of 1 000 mAh·g-1at a current density of 200 mA·g-1.In this case,the stable cycling can still last 32 cycles(Fig.S3).It should noted that the electrode has a relatively high catalyst loading of about 1.4 mg·cm-2.The cycling performance of the cell is better than or comparable with those with similar catalysts and same limited capacity[56-57].The better cycle performance of the Li-O2cell with Co3O4@δ-MnO2/Pt catalyst can be attributed to the unique microstructure and components of the cathode.Pt nanoparticles are highly efficient in catalyzing the ORR/OER in the air cathode[58],leading to enhanced formation/decomposition of Li2O2and thereby long cycle life.In addition,the binder-free electrode design also contributes to good cycling stability of the cell with Co3O4@δ-MnO2/Pt catalyst.Of note is that,the cell shows performance degradation after 60 cycles.This may result from factors such as decomposition of electrolyte or Li corrosion.A previous report show that Pt shows no selectivity in its catalytic activity toward Li2O2oxidationreaction and electrolyte decomposition[31],which is also partly responsible for the limited cycle life.
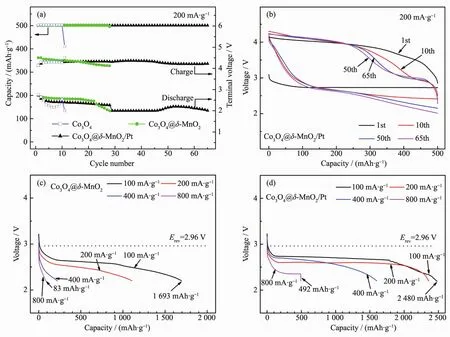
Fig.4 (a)Terminal voltages of Co3O4,Co3O4@δ-MnO2andCo3O4@δ-MnO2/Pt-catalyzed Li-O2cells at a limited capacity of 500 mAh·g-1;(b)Voltage profiles of Co3O4@δ-MnO2/Pt-catalyzed Li-O2cell;Discharge profiles of(c)Co3O4@δ-MnO2 and(d)Co3O4@δ-MnO2/Pt-catalyzed Li-O2cells at various current densities
Fig.4(c,d)show the discharge profiles of the Co3O4@δ-MnO2and Co3O4@δ-MnO2/Pt-catalyzed Li-O2cells at various current densities.In the figures,Erevis the reversible potential of Li-O2cells,namely,2.96 V vs Li/Li+.At 100 mA·g-1,the cell with Co3O4@δ-MnO2/Pt yields a capacity of 2 480 mAh·g-1and exhibits a high discharge plateau of 2.71 V.When the current density increases from 100 to 800 mA·g-1,the discharge capacity and plateaus decrease gradually.But even at 800 mA·g-1,the cell could still deliver a capacity of 492 mAh·g-1due to the excellent catalytic activity offor ORR.In comparison,the cell withcatalyst shows a lower capacity of 1 693 mAh·g-1and a lower discharge plateau of 2.59 V at 100 mA·g-1.The discharge capacity decreases rapidly to 83 mAh·g-1when the current density increases to 800 mA·g-1.The low capacity of the Co3O4@δ-MnO2-catalyzed cell indicates its poor ORR catalytic activity.The obviously enhanced catalytic activity of Co3O4@δ-MnO2/Pt is closely related to the introduction of Pt.It is expected that the presence of Pt will alter the crystallization behavior of Li2O2by supplying catalytically active sites.
To clarify the superior catalytic activity of Co3O4@δ-MnO2/Pt and the role that it plays in increasing the cycle performance of the cell,the electrodes after discharge were observed by SEM and TEM.The loading of the SEM/TEM holders to the chamber was finished as soon as possible to minimize the exposure of the samples to air.As shown in Fig.5(a,b),no large Li2O2particles can be found on the discharged Co3O4electrode,and the pristine array-type structure ofwas generally preserved after discharge to 500 mAh·g-1.To reveal the growth position of Li2O2on the electrode,the morphologies were observed and compared to the original Co3O4@δ-MnO2/Pt.In the discharged electrodes,it seems that the Li2O2forms inside the porous δ-MnO2and a fluffy substance grows conformally along the surface of thearrays,where the original array structure with voids is clearly visible.Namely,the pores in the pristinewere filled by the Li2O2.This form of Li2O2is usually rich in defects and poorly crystallized and thus easily to be decomposed[59].The fluffy substance is further characterized by TEM (Fig.5c)and is confirmed to be Li2O2by selected area electron diffraction(SAED,Fig.5d).From these results,it can be concluded thatPt arrays catalyzes the conformal growth of thinlayered Li2O2along the surface of electrode.
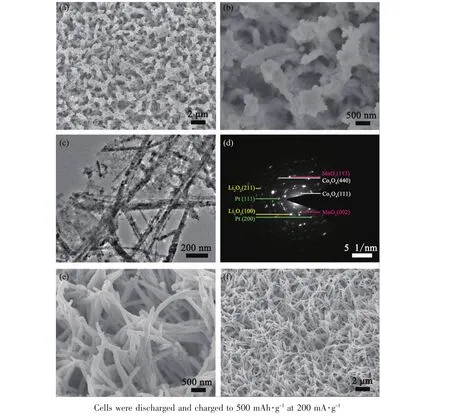
Fig.5 (a,b)SEM images,(c)TEM image and(d)SAED pattern of Co3O4@δ-MnO2/Pt after the first discharge;(e,f)SEM images of Co3O4@δ-MnO2/Pt after the first charge
For theelectrode, after recharge,the porous structure is visible again,indicative of sufficient decomposition of the discharge product(Fig.5e).Besides,the array-type structure of Co3O4@δ-MnO2/Pt remains intact(Fig.5f).The reversible formation/decomposition of Li2O2is confirmed by Li1s XPS (Fig.6a)and further supported by EIS measurements(Fig.6b and Table 1).In the Nyquist plots,the fitted curves are obtained by using the equivalent circuit in the inset,where Rerepresents ohm resistance of cell components,Rfand Q1represent surface film resistance and relaxation capacitance,Rctand Q2correspond to the charge transfer resistance and double-layer capacitance,and Zwis associated with the bulk diffusion of Li ions.In the table,Y is admittance response of constant phase element Q1and Q2and n is index of the angular frequency[60].The remarkable increase in Rct(from 331.9 to 767.1 Ω)after discharge indicates the deposition of insulating Li2O2which passivates the electrode,whereas the decrease in Rct(from 767.1 to 301.6 Ω)denotes the sufficient removal of Li2O2after recharge.From these results,it can be seen that the Ni-supported Co3O4@δ-MnO2/Pt electrode is highly efficient in catalyzing ORR/OER by controlling the Li2O2growth,which can explain the high discharge capacity and long cycle life of the cells.
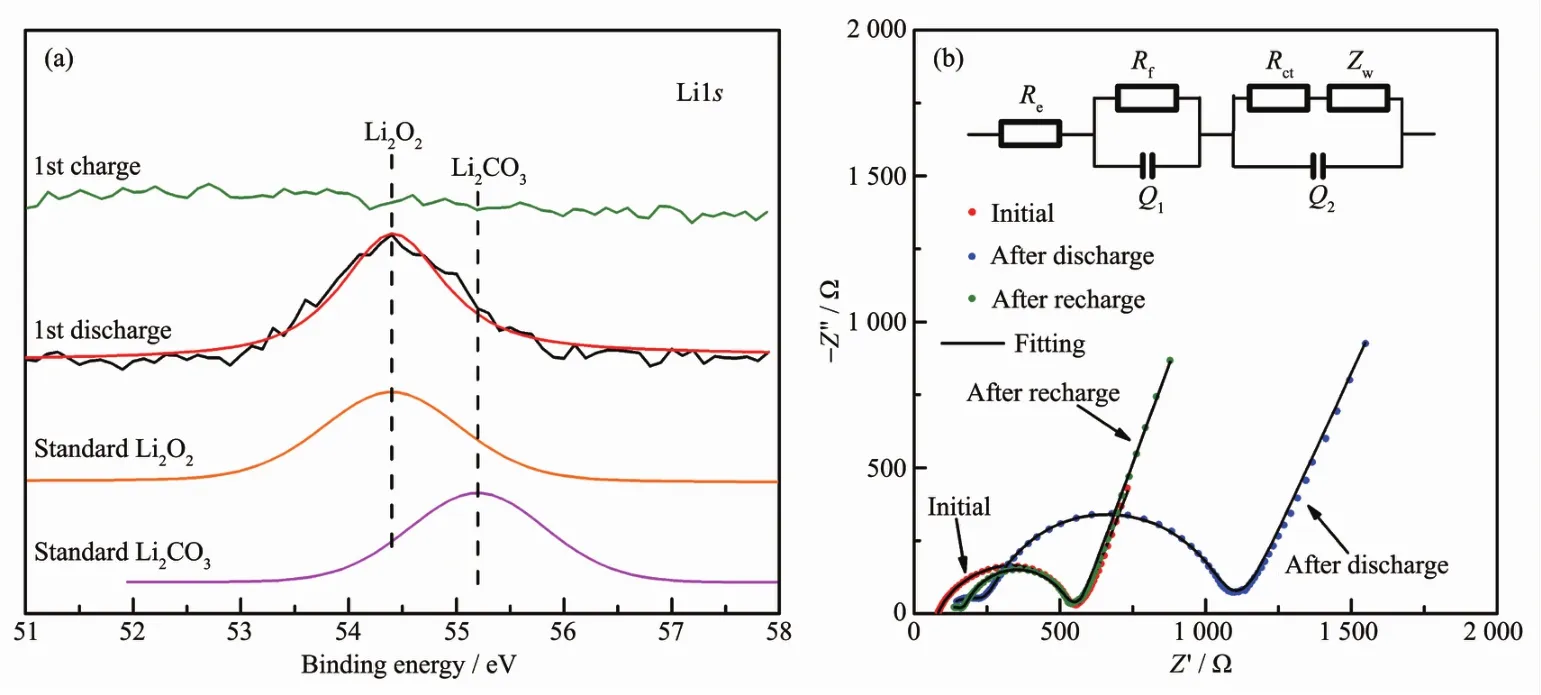
Fig.6 (a)Li1s XPS after the first cycle and(b)Nyquist plots and corresponding fittings of Li-O2cells with Co3O4@δ-MnO2/Pt electrode at the initial state and discharged/recharged to 2.2 V/4.3 V

Table 1 Fitting results of the Nyquist plots using the equivalent circuit
3 Conclusions
In summary,we propose a unique design of a core-shellarrays-type electrode with a controllable,facile route.In this design,the arraytype structure facilitates the electrode wetting and oxygen gas transport and supplies free volume for Li2O2loading.The presence ofPtsuppliesthe catalytically active centers and guides the conformal growth of thin-layered Li2O2.The conformal,thinlayered growth of Li2O2on Co3O4@δ-MnO2/Pt enables its easy decomposition upon charge.The porous structure of δ-MnO2makes high Li2O2loading possible even though it has a conformal growth mode.As a result,Li-O2cell catalyzed by Co3O4@δ-MnO2/Pt arrays delivers high discharge capacity (2 480 mAh·g-1at 100 mA·g-1)and shows good cycling stability(65 cycles at 200 mA·g-1with a limited capacity of 500 mAh·g-1).This work provides a new design of efficient catalytic cathode for Li-O2cells.
Supporting information is available at http://www.wjhxxb.cn
[1]Abraham K M,Jiang Z.J.Electrochem.Soc.,1996,143(1):1-5
[2]Ogasawara T,Débart A,Holzapfel M,et al.J.Am.Chem.Soc.,2006,128(4):1390-1393
[3]Girishkumar G,McCloskey B,Luntz A C,et al.J.Phys.Chem.Lett.,2010,1(14):2193-2203
[4]Park M,Sun H,Lee H,et al.Adv.Energy Mater.,2012,2(7):780-800
[5]Bruce P G,Freunberger S A,Hardwick L J,et al.Nat.Mater.,2012,11(1):19-29
[6]Luntz A C,McCloskey B D.Chem.Rev.,2014,114(23):11721-11750
[7]Aurbach D,McCloskey B D,Nazar L F,et al.Nat.Energy,2016,1:16128
[8]Geng D S,Ding N,Hor T S A,et al.Adv.Energy Mater.,2016,6(9):UNSP 1502164
[9]Yi J,Guo S H,He P,et al.Energy Environ.Sci.,2017,10(4):860-884
[10]Feng N N,He P,Zhou H S.Adv.Energy Mater.,2016,6(9):1502303
[11]Viswanathan V,Thygesen K S,Hummelshj J S,et al.J.Chem.Phys.,2011,135(21):214704
[12]Gerbig O,Merkle R,Maier J.Adv.Mater.,2013,25(22):3129-3133
[13]Shao Y Y,Ding F,Xiao J,et al.Adv.Funct.Mater.,2013,23(8):987-1004
[14]YANG Feng-Yu(杨凤玉),ZHANG Lei-Lei(张蕾蕾),XU Jie-Jing(徐吉静),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(8):1563-1573
[15]Chang Z W,Xu J J,Liu Q C,et al.Adv.Energy Mater.,2015,5(21):1500633
[16]Ma Z,Yuan X X,Li L,et al.Energy Environ.Sci.,2015,8(8):2144-2198
[17]Wen Z Y,Shen C,Lu Y.ChemPlusChem,2015,80(2):270-287
[18]Mitchell R R,Gallant B M,Thompson C V,et al.Energy Environ.Sci.,2011,4(8):2952-2958
[19]Jung H G,Hassoun J,Park J B,et al.Nat.Chem.,2012,4(7):579-585
[20]Zhang M,Xu Q,Sang L,et al.Chin.Sci.Bull.,2014,59(24):2973-2979
[21]Guo Z Y,Zhou D D,Dong X L,et al.Adv.Mater.,2013,25(39):5668-5672
[22]Yu M Z,Zhou S,Liu Y,et al.Sci.China Mater.,2017,60(5):415-426
[23]Liu T,Leskes M,Yu W J,et al.Science,2015,350(6260):530-533
[24]McCloskey B D,Speidel A,Scheffler R,et al.J.Phys.Chem.Lett.,2012,3(8):997-1001
[25]Ottakam Thotiyl M M,Freunberger S A,Peng Z Q,et al.J.Am.Chem.Soc.,2013,135(1):494-500
[26]Lu Y C,Xu Z C,Gasteiger H A,et al.J.Am.Chem.Soc.,2010,132(35):12170-12171
[27]Peng Z Q,Freunberger S A,Chen Y H,et al.Science,2012,337(6094):563-566
[28]Xu J J,Wang Z L,Xu D,et al.Nat.Commun.,2013,4:2438(10 Pages)
[29]Li C C,Zhang W Y,Ang H X,et al.J.Mater.Chem.A,2014,2(27):10676-10681
[30]Sun B,Chen S Q,Liu H,et al.Adv.Funct.Mater.,2015,25(28):4436-4444
[31]Jeong Y S,Park J B,Jung H G,et al.Nano Lett.,2015,15(7):4261-4268
[32]Luo W B,Gao X W,Chou S L,et al.Adv.Mater.,2015,27(43):6862-6869
[33]Jiang J,He P,Tong S F,et al.NPG Asia Mater.,2016,8:e239(7 Pages)
[34]Cao Y,Wei Z K,He J,et al.Energy Environ.Sci.,2012,5(12):9765-9768
[35]Hu X F,Han X P,Hu Y X,et al.Nanoscale,2014,6(7):3522-3525
[36]Hu Y X,Zhang T R,Cheng F Y,et al.Angew.Chem.Int.Ed.,2015,54(14):4338-4343
[37]Cui Y M,Wen Z Y,Liu Y.Energy Environ.Sci.,2011,4(11):4727-4734
[38]Black R,Lee J H,Adams B,et al.Angew.Chem.Int.Ed.,2013,52(1):392-396
[39]Wang S F,Sha Y J,Zhu Y L,et al.J.Mater.Chem.A,2015,3(31):16132-16141
[40]Tong S F,Zheng M B,Lu Y,et al.J.Mater.Chem.A,2015,3(31):16177-16182
[41]Liu W M,Gao T T,Yang Y,et al.Phys.Chem.Chem.Phys.,2013,15(38):15806-15810
[42]CAI Sheng-Rong(蔡生容),WANG Xiao-Fei(王晓飞),ZHU Ding(朱丁),et al.Chinese J.Inorg.Chem.(无机化学学报),2016,32(12):2082-2087
[43]Chang Y Q,Dong S M,Ju Y H,et al.Adv.Sci.,2015,2(8):1500092
[44]Liu Q C,Xu J J,Xu D,et al.Nat.Commun.,2015,6:7892(8 Pages)
[45]Zhao G Y,Mo R W,Wang B Y,et al.Chem.Mater.,2014,26(8):2551-2556
[46]Kim S T,Choi N S,Park S,et al.Adv.Energy Mater.,2015,5(3):1401030
[47]Aetukuri N B,McCloskey B D,García J M,et al.Nat.Chem.,2015,7(1):50-56
[48]Lau S,Archer L A.Nano Lett.,2015,15(9):5995-6002
[49]Radina M D,Siegel D J.Energy Environ.Sci.,2013,6(8):2370-2379
[50]Wang J W,Zhang Y L,Guo L M,et al.Angew.Chem.Int.Ed.,2016,55(17):1-6
[51]Qin Y,Lu J,Du P,et al.Energy Environ.Sci.,2013,6(2):519-531
[52]Wang G Q,Tu F F,Xie J,et al.Adv.Sci.,2016,3(10):1500339
[53]He G Q,Song Y,Liu K,et al.ACS Catal.,2013,3(5):831-838
[54]Song W Q,Poyraz A S,Meng Y T,et al.Chem.Mater.,2014,26(15):4629-4639
[55]Trahey L,Karan N K,Chan M K Y,et al.Adv.Energy Mater.,2013,3(1):75-84
[56]Cao J Y,Liu S Y,Xie J,et al.ACS Catal.,2015,5(1):241-245
[57]Wu F,Zhang X X,Zhao T L,et al.J.Mater.Chem.A,2015,3(34):17620-17626
[58]Zahoor A,Christy M,Kim Y,et al.J.Solid State Electrochem.,2016,20(5):1397-1404
[59]HUANG Jun(黄俊),PENG Zhang-Quan(彭章泉).Energy Storage Science and Technology(储能科学与技术),2018,7(2):167-174
[60]Piao T,Park S M,Doh C H,et al.J.Electrochem.Soc.,2009,146(8):2794-2798
