Transcriptomes of early developing tassels under drought stress reveal differential expression of genes related to drought tolerance in maize
2018-06-06WANGNanLlLiangGAOWenweiWUYongboYONGHongjunWENGJianfengLlMingshunZHANGDeguiHAOZhuanfangLlXinhai
WANG Nan, Ll Liang , GAO Wen-wei, WU Yong-bo YONG Hong-jun WENG Jian-feng Ll Mingshun ZHANG De-gui HAO Zhuan-fang , Ll Xin-hai
1 Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, P.R.China
2 College of Agriculture, Xinjiang Agricultural University, Urumqi 830000, P.R.China
1. lntroduction
Drought is one of the major environmental stresses affecting maize production, and inadequate rainfall, due to climate change, will negatively impact agricultural productivity. The improvement of maize drought tolerance has long been important for enhancing the stability of food production,although conventional breeding most often takes advantage of favorable genetic recombination among germplasm to improve yield. The comprehensive elucidation of molecular regulatory mechanisms by which maize responds to drought stress is fundamentally important for maize genetics and breeding (Xionget al. 2002; Nakashimaet al. 2014).Abiotic stresses including drought, high temperature, and salinity all influence the expression of a large number of genes that induce or regulate other genes through complex transcriptional networks (Todakaet al. 2015).Transcriptome analyses of plant tissues and organs under drought-stressed conditions will reveal relevant genes and pathways and thereby helping unravel the mechanisms of drought tolerance.
Before, some drought-inducible genes have been identified by analyzing the transcriptomes of maize reproductive organs using microarray technology (Zhuanget al. 2007). These genes encode cell stress-protective proteins and regulatory proteins, as well as signal transduction components (Shinozakiet al. 2003). Among techniques available for transcriptome analysis, microarrays require the design and synthesis of probes for known genes.This method has some technical limitations, including false positives generated by hybridization of multiple gene products to the arrays, which has limited their usefulness(Okoniewski and Miller 2006; Shenet al. 2014). Recently,along with the rapid and economic sequencing technology,sequencing method based on RNA level and its research results have systematically altered our view of the extent and complexity of eukaryotic transcriptomes (Wanget al.2009). A total of 619 maize drought response genes were identified using RNA-Seq, and 20 genes regulating flowering time were further analyzed, such asPRR37andCONSTANS(Leeet al. 2017). Drought-induced abortion of maize embryos suggested that signaling events such as increased abscisic acid (ABA) levels and decreased glucose levels interfered with ABA and sugar signaling pathways, which might have activated programmed cell death or senescence and arrested the cell cycle in the developing maize ovary(Kakumanuet al. 2012).
Drought or heat severely affects normal pollination and decreases yield in maize. The period from meiosis to tetrad break-up in anthers and anthesis are key sensitivity periods to drought for maize. Drought stress during the first stage inhibits development of microspores or pollen, then causing male sterility (Saini 1997). Zhuanget al. (2007) identified 1 513 transcripts that were differentially expressed during meiosis in immature tassels and ears under drought-stressed and well-watered conditions, and 33 up-regulated transcripts were identified in both immature ears and tassels after drought stress using oligo microarray analysis. However, the metabolic pathways involved and the reasons that drought stress results in male sterility are still not clearly understood. In the present study, we used RNA-Seq to explore drought-induced gene expression in early developing tassels from 10 inbred lines,which selected from five heterotic groups of China (Liuet al.2015) with different drought resistance (Haoet al. 2011), under both well-watered and drought-stressed conditions. The goal of the present study was: i) to identify transcripts that were differentially expressed under drought-stress condition, ii) to explore the important gene networks and metabolic pathways induced by drought in early developing tassels, and iii) to propose transcriptional and other mechanisms of droughtinduced male sterility in maize.
2. Materials and methods
2.1. Plant materials and watering treatments
The 10 inbred maize (Zea maysL.) lines were planted in 10-L pots containing a 2:2:1:1 (v/v) mix of field soil:peat:fowl manure:perlite with 1 g of fritted trace elements (Table 1).More than 20 parallel pots of each inbred line were planted for getting the same size of developing tassels after peeling off in the summer of 2014. Plants were irrigated by hand every other day to maintain soil water content close to capacity after the 3-leaf stage, and pesticides were applied every 2 weeks. When the plants reached to 9- or 10-leaf (V9 or V10)stage, some of them were subjected to drought-stressed condition, and some were still under well-watered condition(control). After treatment, when plants reached to floralorgan differentiation stage (V11 to V13 stages, varied among inbreds), early developing young tassels were sampled for RNA-Seq. At the same time, soil moisture content (%) was recorded to monitor the level of drought stress applied to each maize line. Steady-state leaf fluorescence was also recorded on leaves to reflect the corresponding response of plants under drought-stressed or well-watered conditions(Mini-PAM, Effeltrich, Germany). When the soil moisture content in drought-stressed treatments fell to about 25%,the sheath leaves of maize were stripped from plants, then the early developing tassels were sampled from all inbred lines in the experiment.

Table 1 The information of 10 inbred lines experimented in this study1)
2.2. lsolation of total RNA from tassel samples and RNA-Seq
A total of 20 tassel samples from each of 10 inbred lines under drought-stressed and well-watered conditions were homogenized in liquid nitrogen. Total RNA was extracted from each sample using TRIzol®Reagent (Invitrogen,USA), according to the manufacturer’s protocol. As all the plants would be identified corresponding drought-tolerant responses under physiological dehydration, we took three inbreds at least as replications to select significantly differentially expressed genes for further analysis.
Total RNA samples were sequenced at Beijing Genomics Institute, Shenzhen, China. The poly-A+RNA was sheared into short fragments of approximately 200 bp, and cleaved RNA fragments were copied into the first-strand of cDNA using random-hexamer primers and reverse transcriptase(Qiagen, Duesseldorf, Germany). The second-strand of cDNA was synthesized in reactions containing DNA polymerase I, RNaseH, buffer, and dNTPs (TransGen Biotech, Beijing, China). The cDNA fragments were purified using AMPure XP beads (Beckman Coulter, USA). Adaptorligated cDNAs were enriched by PCR in reactions to create the final cDNA library. Libraries were sequenced on the Illumina HiSeq 2000 Platform (Illumina, USA) according to the manufacturer’s recommendations.
2.3. Data processing and analysis
Raw reads were subjected to quality control, and the lowquality reads (low-quality bases in raw reads of >30%)and reads containing vector or adapter sequences were removed before data analysis. Clean reads acquired were aligned to the maize B73 reference genome (AGPv2) using SOAPaligner/SOAP2 (www.maizesequence.org) (Liet al.2009), only allowing mismatches of less than five bases between alignments. Digital gene expression counts were normalized by log2transformation and compared using the reads per kilobase transcriptome per million mapped reads method (RPKM) (Mortazaviet al. 2008). The specific algorithm developed by BGI for calculating gene expression was showed previously in Fuet al. (2013). Comparative analysis of differentially expressed genes (DEGs) from 10 inbred lines was performed using Microsoft Office 2013(Microsoft Corporation, US). Here, an false discovery rate(FDR)≤0.001 (Benjamini and Yekutieli 2001), an absolute value of the Log2Ratio≥1, andP≤0.001 were used as the thresholds by which to assess the significance of differences in gene expression.
All DEGs were mapped to Gene Ontology (GO) terms under the top-level terms or domains at the GO database(http://www.geneontology.org), and transcript numbers were calculated for each term. Then a hypergeometric test with a Bonferroni correction for multiple comparisons was used to identify significantly enriched GO terms in the input list of DEGs using GO::Term Finder with a threshold ofP≤0.05 (Abdi 2007).Kyoto encyclopedia of genes and genomes (KEGG) was used to identify metabolic or signal transduction pathways that were significantly enriched between DEGs sampled from droughtstressed or well-watered plants relative to the transcriptomic background of early developing tassels (Kanehisaet al. 2008).
2.4. Verification of transcript expression using qRT-PCR
Seeds of the inbred line Qi 319 were germinated on moist filter paper in an incubator (28°C/22°C for 14 h/10 h). After four days, the germinated seedlings were then planted in quartz sand in a greenhouse. The seedling endosperms were peeled and transferred to Hoagland’s nutrient solution at 2-leaf (V2) stage. At 3-leaf (V3) stage, the seedlings were treated with 20% polyethylene glycol 6 000 (PEG-6000).
Total RNA was extracted using Trizol (Transgen Biotech,Beijing, China) from leaves, stems, and roots of seedling after 0 (control), 1, 3, 6, and 12 h of PEG treatment. qRTPCR was performed using SuperReal PreMix Plus (SYBR Green) Kit (Tiangen) and products were quantified using a iQ5/MyiQ2 Real-time PCR Detection System (Bio-Rad,California, USA). Expression of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene from maize was used as an internal control for gene expression. Relative gene expression was calculated using the 2–ΔΔCTmethod(Livak and Schmittgen 2001).
3. Results
3.1. Phenotypic and photosynthetic responses to drought stress
After five days without watering at floral organ differentiation stage of young tassel in the drought-stressed treatment, soil moisture contents in the pots decreased to an average of 24.08% with the highest of 26.80% in Chang 7-2 and the lowest of 22.00%in X178. In contrast, soil moisture contents in well-watered treatment remained constantly above 62.00%, with the highest of 64.30% in Qi 319 and the lowest of 62.00% in Dan 340, indicating that the control was in well-watered condition (Table 2).Because plant development is dynamic, the steadystate chlorophyll fluorescence of the uppermost leaf of each line was also monitored to determine the degree of photosynthetic inhibition due to drought stress. Average steady-state leaf fluorescence decreased by 0.127 under drought stress compared with that of control. As predicted, the tassel lengths sampled from all plants under drought-stressed condition were smaller than those under well-water condition (control). On average, the tassel length decreased from 4.11 to 3.01 cm after drought stress(Table 2 and Fig. 1). These results indicated that tassel growth was inhibited and gene expression was changed under drought stress.
3.2. Maize transcriptome revealed gene expression changes under drough-stress condition
A total of 940 million high-quality reads were generated from the transcriptomes of 20 immature tassels, which derived from 10 maize inbred lines under control and drought-stressed conditions.About 76.88% of the reads were mapped to the B73 reference genome (AGPv2) and 78.42%were mapped to the annotated maize genes,which revealed that there were still 23.12% unique sequences (50 487 novel transcripts) in these lines when compared with that of B73. Among these annotated genes, 76.16% genes covered >50% of the entire gene (Fig. 2). An average of 78.42% of genes could be uniquely mapped and 29 582 expressed genes were identified. After filtering, a total of 19 028 significant DEGs (FDR≤0.001, Log2Ratio≥1,andP≤0.001) were identified from each line as enriched in some metabolic pathways under droughtstressed conditions. Of these, 8 518 differentially expressed genes were up-regulated and 10 510 genes were down-regulated (Table 3). Interestingly,the more drought-tolerant genotypes were, the more up-regulated genes were. For example, the downregulated genes were 8.05 and 1.94 times than the up-regulated genes in drought-tolerant inbreds Qi 319 and Tie 7922, respectively, but 0.26 and 0.92 times in drought-susceptible inbreds CA339 and Ji 81162, respectively.


Fig. 1 The sampled young tassels of 10 inbred lines. The left and right tassels of each line was under drought-stressed and wellwatered conditions (control), respectively.

Fig. 2 An average of gene coverage percent by reads.
The significant pathways identified in at least three inbred lines were further summarized in Table 4. RNA transport and mRNA surveillance were the most two pathways enriching in genes with binding functions, and were highly expressed in plant tassels under drought-strssed condition, except for CA339 with 23.62% of the enriched transcripts. Droughttolerant line Tie 7922 had the largest proportion (35.77%) of genes encoding proteins involved in RNA transport in droughtstressed tassels. The following enriched metabolic pathways were biosynthesis of secondary metabolites, amino acids,and macromolecular metabolites such as carbohydrates,lipids, glycans, terpenoids. Among these, pathways for secondary metabolites showed the greatest change in DEG profiles under drought stress, and genes encoding enzymes involved in the biosynthesis of these compounds accounted for greater than 11.34% of all DEGs in six inbreds, including Qi 319, X178, Ye 478, B73, Dan 340, and Ji 81162. Additionally,genes encoding enzymes involved in ‘phenylpropanoid and stilbenoid biosynthesis’ and ‘diarylheptanoid and gingerol biosynthesis’ in pathways for secondary metabolites biosynthesis were also up-regulated during drought response in early developing tassels (Table 4 and Fig. 3). Moreover,primary metabolic pathway categories affected by drought stress included genes expression for carbohydrate and lipid metabolism. Greater than 1.36% of all DEGs in six inbred lines (X178, Yu 12, Chang 7-2, Ye 478, B73, and Dan 340)corresponded to genes encoding enzymes were involved in galactose metabolism, and greater than 0.37% of all DEGs in five inbred lines (Qi 319, X178, Ye 478, B73, and Ji 81162)corresponded to genes encoding enzymes involved in fatty acid elongation (Table 4 and Fig. 3).
3.3. GO term enrichment of the drought-stressed maize transcriptome
The frequencies of DEGs under the top three most significantly enriched GO terms are shown in Fig. 4. In the‘molecular function’ category, most of the DEGs belonged to the ‘binding’ and ‘catalytic activity’ categories. Other terms such as ‘antioxidant activity’, ‘electron carrier activity’ and‘enzyme regulator activity’ represented an average of less than 10% of the DEGs affected by drought. For the ‘cellular component’ domain, the GO terms ‘cell’, ‘cell part’ and‘organelle’ represented over 60% of DEGs, indicating that drought stress had a great impact on cellular function. And we found that three membrane-related GO terms, were also enriched in our study, as Osakabeet al. (2014) showed that membrane transport played an important role in maintaining cellular homeostasis under drought stress. In the ‘biological process’ domain, 22 GO terms were enriched. Among these, except for ‘metabolic process’ and ‘cellular process’,comprising more than half of the DEGs, were affected by drought stress, some other biological processes, such as‘single-organism process’, and ‘response to stimulus’, were also affected by drought stress to some degree.

Table 3 Number of differentially expressed genes in 10 maize inbred lines under drought-stressed condition

Table 4 Drought-induced pathways and related differentially expressed gene with annotation in 10 inbred lines
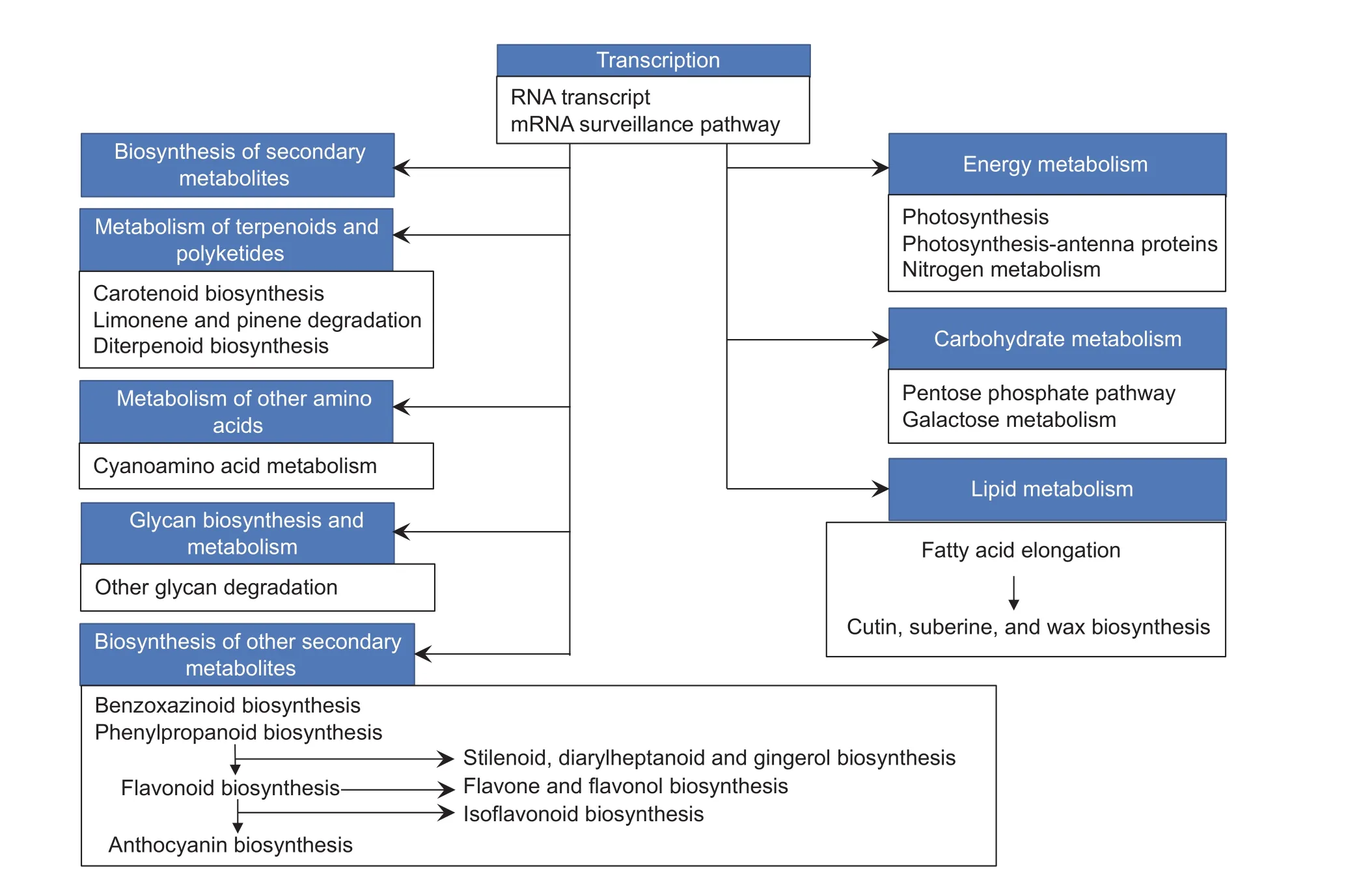
Fig. 3 The major response metabolisms under drought-stressed in young tassel derived from RNA-Seq. The three or more than three significantly common pathways in 10 inbred lines under drought stress were collected in black rectangles. These branch pathways was a portion of backbone metabolisms in blue boxes.

Fig. 4 Main enrichment analysis of Gene Ontology (GO) annotation of differentially expressed genes (DEGs) in cellular component,molecular function and biological process. At least three tassels for each of inbred line with common GO annotations under drought stress were collected, the horizontal axis represents the percentages of DEGs involved in one GO term. The error bars show standard deviations for different lines.
3.4. ldentification of DEGs under drought stress in developing maize tassels
The largest proportion of DEGs from drought-stressed tassels was assigned to the KEGG pathways (Table 4).Of them, the exon-junction complex (EJC)-related genes were an important part of the ‘RNA transcript’ and ‘mRNA surveillance’ pathways that influence translation (Tangeet al. 2004). In this study, almost all annotated DEGs in the ‘mRNA surveillance pathway’ were enriched for genes encoding two components of the EJC, RNPS1 (RNA-binding protein with serine-rich domain 1) and SRm160 (Serine/arginine repetitive matrix protein 1). A total of 87 DEGs related to RNPS1 and SRm160 were identified in droughtstressed tassels in five or more inbred lines (Appendix A).This pathway annotation indicated that in drought-stressed young tassels, the EJC could regulate splicing, MAPK signaling, and transport by affecting the expression of genes encoding RNPS1 and SRm160.
Nineteen DEGs representing genes encoding putative transcription factors were also noted in drought-stressed young tassels, including zinc finger proteins (4), bHLH(4), NAC (3), WRKY (2), bZIP (1), HB (1), SPL (1), LLY(1), Trihelix (1), and GRF (1). Genes encoding three transcription factors (hb8, GRMZM2G135447; c3h39,GRMZM5G801627; and dof26, GRMZM2G061292) showed increased expression in drought-stressed young tassels in at least four inbred lines. The expression ofdof26respondses to salt stress in maize seedlings (Chen and Cao 2015).However, the transcript abundance of genes encoding three members of the abscisic acid-, stress-, and ripening-induced(ASR)gene family (ZmASR1, GRMZM2G136910; ZmASR2,GRMZM5G854138; and ZmASR5, GRMZM2G052100),decreased in early developing tassels from most of inbred lines under drought stress. Transcripts of these genes accumulated in response to environmental stresses and might have effects in protecting cellular components and regulating gene expression (Perez-Diazet al. 2014). The exact regulation mechanism of these DEGs should be further investigated.
3.5. Verification and expression of regulative DEGs in leaf, stem and root under drought stress
For verification of transcription factors expressed in drought-stressed tassels, three of them,hb8,c3h39anddof26, were further tested with the inbred Qi 319 (Fig. 5).After treating with PEG-6000, all the three genes showed down-expressed in roots after 12 h treatment. Interestingly,c3h39anddof26showed nearly the same expression patterns in all treated tissues, which expression increased gradually, peaked at 3 h PEG treatment, then declined in stem. But the expression ofhb8was a little earlier than other genes, with the highest level at 1 h PEG treatment in all treated tissues, then it rapidly declined in roots and stems and remained higher expression level in leaves.
4. Discussion
4.1. The transcriptome of maize early developing tassels changes significantly under drought stress
The floral organ differentiation stage is important for tassel and anther development. Except RNA regulation at the transcriptional level played important role in drought-stressed response in maize, a large number of DEGs encoding proteins involved in several metabolic pathways related to drought stress response were identified in early developing maize tassels under drought stress. Although gene expression profiles were highly variable among inbred lines, common and important regulation genes and their underlying pathways were identified in at least three inbreds. As reported in other species, the major metabolic pathways that were identified in tassels under drought stress might play important roles in the adaptation of drought, especially in the late period of drought stress. In drought-stressed tobacco root,transcripts of genes encoding enzymes involved in fatty acid,carbohydrate, and lipid metabolism-related pathways, as well as oxidoreductases, were greatly enriched, and a total of 17 pathways were significantly affected by drought stress (Yinet al. 2015). In diploidPaulowina australis, ‘photosynthesis’,‘biosynthesis of secondary metabolites’, and ‘phenylpropanoid biosynthesis’ pathways were significantly enriched in response to drought (Donget al. 2014). Transcriptome sequencing of moso bamboo (Phyllostachys edulis) at different flowering stages showed that transcripts related to‘genetic information processing’ were the most abundant,including those in ‘RNA transcript and mRNA surveillance’pathway (Gaoet al. 2014). In developing rice embryos, DEGs in pathways for carbohydrate, lipid, and energy metabolism were enriched, including those encoding enzymes involved in starch and sucrose metabolism, glycolysis, and fatty acid metabolism (Xuet al. 2012).
Anthocyanin biosynthesis was related to steadystate leaf fluorescence of maize. In this study, two genes (GRMZM2G106443 and GRMZM5G896260),encoding glycosyltransferase and involved in anthocyanin biosynthesis, were down-regulated in Yu 12, which might keep the steady-state leaf fluorescence of Yu 12 after drought stress. GRMZM5G896260 was also down-regulated in Tie 7922 with relatively slightly decreased steady-state leaf fluorescence. These results suggested that the differential expression of genes encoding proteins involved in the regulation of RNA behavior, the biosynthesis of carbohydrates and lipids, and energy metabolism could likely be important for plant development and drought adaption.
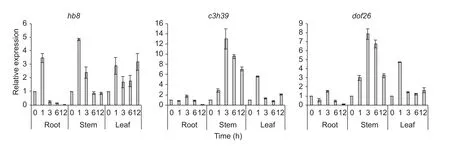
Fig. 5 Changes in transcript levels of three transcription factors under drought stress. Error bars represented standard deviation for the three biologic replicates of qRT-PCR experiments.
4.2. Responses of genes encoding enzymes involved in carbohydrate metabolism to drought stress
A large number of DEGs related to galactose metabolism were observed in drought-stressed tassels of six inbred lines (B73, Chang 7-2, Dan 340, X178, Ye 478, and Yu 12)(Appendix B and Fig. 6). Among these DEGs, the transcript abundance of gene (GRMZM2G052336), which encoded UDP-glucose 4-epimerase, decreased in drought-stressed tassels in four lines (B73, Dan 340, Ye 478, and Yu 12).Genes encoding invertase and alpha-glucosidase (bins A4 and A5 of Fig. 6, Appendix B), could break sucrose down into glucose and fructose (Liet al. 2011) and were down-regulated in drought-stressed tassels. Invertase activity is very important in early kernel development for converting sucrose originating from source leaves into hexoses to support endosperm and embryo cell division(Bateet al. 2004). Andersenet al. (2002) suggested that low acid invertase activity could decrease starch biosynthesis and lead to depletion of starch reserves.Enzyme 6-phosphofructokinase, involved in the regulation of glycolysis in eukaryotes, can phosphorylate fructose 6-phosphate to fructose-1,6-bisphosphate with ATP (Nariya and Inouye 2002). In our study, a large proportion of genes that encode 6-phosphofructokinase showed decreased transcript abundance, which could affect glucose and energy metabolism in drought-stressed tassels (bin A6 of Fig. 6,Appendix B). DEGs encoding 6-phosphofructokinase and invertase, includingMin1,Ivr2, and GRMZM2G095725(similar to cell wall invertase 2) were significantly downregulated in drought-stressed developing tassels. These results indicated that invertase activity might result in depleted glucose levels and ATP supplies, which might be lethal to tassels or anthers under drought.

Fig. 6 Effects of drought stress on galactose metabolism. Drought stress responses associated with galactose metabolism in young tassel were shown, with a sucrose and glycolysis metabolism procedure in gray and green box, respectively. A1, hexose-1P-uridylyltransferase; A2, raffinose synthase; A3, α-D-galactosidase; A4, invertase; A5, glucosidase; A6, 6-phosphofructokinase.
4.3. Responses of genes encoding enzymes involved in lipid metabolism to drought stress
Lipid biosynthesis in maize embryo uses hexoses produced by invertases and reversible sucrose synthesis (Liet al.2011), which suggests that changes in expression of genes encoding invertases might also affect lipid metabolism. In our study, decreases in the expression of a large number of genes related to lipid metabolism were observed in early developing tassels under drought stress, including those in pathways for ‘fatty acid elongation’ and ‘cutin, suberin,and wax biosynthesis’ (Fig. 7, Table 5). The expression of genes encoding 3-ketoacyl-CoA synthase, the first step in fatty acid elongation that is important in long-chain fatty acid biosynthesis, decreased significantly in the early developing tassels of inbred lines B73, Ji 81162, Qi 319, X178, and Ye 478 under drought stress (bin A of Fig. 7). Verylong-chain fatty acids are precursors for cuticular wax biosynthesis activated by fatty acid synthesis and elongation in the endoplasmic reticulum by enzymes including three ketoacyl-CoA synthase (Lokeshet al. 2013). The fatty acid elongase function of 3-ketoacyl-CoA synthase determines fatty acid chain length and controls synthesis of subsequent elongated products including ketones and wax esters(Lokeshet al. 2013). Mutants such asCER1orWAX2in wax biosynthesis pathway genes generate conditional male sterile or glossy stem phenotypes (Aartset al. 1995;Wanget al. 2015).

Fig. 7 The major genes to response drought in lipid metabolism. Drought stress responses associated with fatty acid elongation(A) and cutin, suberin and wax biosynthesis (B1, B2, C1, C2, and C3) in lipid metabolism were shown, with a table showing gene ID, annotation functions and fold change values of each line. The values in red and green indicated fold increase and decrease in expression of different lines in drought-induced tassels, respectively. The boxes in grayness indicated that the values of mapped reads in well-watered (control) and drought-stressed were less than 30. The N in green indicated that the values of expression in drought-stressed were zero.

Table 5 Key fatty acid elongation genes and wax biosynthesis response to drought stress in young tassel
Two DEGs, representing members of the cytochrome P-450 omega hydroxylase superfamily (GRMZM2G323830 and GRMZM2G022996) that encoding fatty acid hydroxylases(Matsunoet al. 2009), decreased in transcript abundance in early developing tassels under drought stress. These enzymes would be involved in ω-hydroxy fatty acid and longchain ketone biosynthesis (bins B1 and C2 of Fig. 7). The transcript abundances of two other genes in this superfamily(GRMZM2G062946 and GRMZM2G400129) increased slightly in line X178 (read values <30 under control and drought-stressed conditions). In addition, the expression of genes (GRMZM2G086210 and GRMZM2G141214)encoding glucose-methanol-choline (GMC) oxidoreductase,which oxidizes aliphatic alcohols to aldehydes during ω-dicarboxylic acid biosynthesis (Etxebesteet al. 2012),decreased significantly in early developing tassels under drought stress. Another fatty acid hydroxylase gene(GRMZM2G066578) also showed decreased expression in the drought-stressed tassels. However, the DEG encoding fatty aldehyde decarbonylase (GRMZM2G029912)showed increased transcript abundance in line X178.Interestingly, the expression of genes (GRMZM2G036217,GRMZM2G120938, andGRMZM2G480516), which encode fatty acyl reductases decreased significantly in early developing tassels under drought stress, which could have affected long-chain wax ester biosynthesis in tassels (bin C3 of Fig. 7). Fatty acyl reductases are essential for pollen development and could be related to male sterility under drought stress (Aartset al. 1997).Overall, these data suggest that the down-regulation under drought stress of the expression of these genes encoding enzymes involved in fatty acid elongation and suberin and wax biosynthetic pathways could have affected pollen development and could have led to male sterility in drought-stressed tassels.
5. Conclusion
Transcriptome of early developing tassel associated with drought tolerance in maize showed that most of the active metabolic pathways occurred at RNA level, especially the regulation of exon-junction complexes, thus we deduced that transcription regulation might play key roles in abiotic tolerance. Moreover, the carbohydrate and lipid metabolisms related to osmotic adjustment under water-stressed condition were involved in, with most encoding enzymes down-regulated. At the same time, a lot of important genes were identified, which would help to elucidate transcription regulation mechanisms related to drought tolerance.
Acknowledgements
This research was jointly funded by the National Natural Science Foundation of China (31661143010) and the Pilot Project of Breeding of the Seven Major Crops, China(2016YFD0101803).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
Aarts M G M, Hodge R, Kalantidis K, Florack D, Wilson Z A,Mulligan B J, Stiekema W J, Scott R, Pereira A. 1997. TheArabidopsisMALE STERILITY 2 protein shares similarity with reductases in elongation/condensation complexes.The Plant Journal,12, 615–623.
Aarts M G M, Keijzer C J, Stiekema W J, Pereira A. 1995.Molecular characterization of theCER1gene ofArabidopsisinvolved in epicuticular wax biosynthesis and pollen fertility.The Plant Cell,7, 2115–2127.
Abdi H. 2007. The bonferonni and Šidák corrections for multiple comparisons. [2017-04-12]. http://www.utdallas.edu/~herve/Abdi-Bonferroni2007-pretty.pdf
Andersen M N, Asch F, Wu Y, Jensen C R, Naested H,Mogensen V O, Koch K E. 2002. Soluble invertase expression is an early target of drought stress during the critical, abortion-sensitive phase of young ovary development in maize.Plant Physiology,130, 591–604.
Bate N J, Niu X P, Wang Y W, Reimann K S, Helentjaris T G. 2004. An invertase inhibitor from maize localizes to the embryo surrounding region during early kernel development.Plant Physiology,134, 246–254.
Benjamini Y, Yekutieli D. 2001. The control of the false discovery rate in multiple testing under dependency.The Annals of Statistics,29, 1165–1188.
Chen Y Z, Cao J. 2015. Comparative analysis of dof transcription factor family in maize.Plant Molecular Biology Reporter,33, 1245–1258.
Dong Y P, Fan G Q, Zhao Z L, Deng M J. 2014. Transcriptome expression profiling in response to drought stress inPaulownia Australis.International Journal of Molecular Sciences,15, 4583–4607.
Etxebeste O, Herrero-Garcia E, Cortese MS, Garzia A,Oiartzabal-Arano E, de los Rios V, Ugalde U, Espeso E A. 2012.GmcAis a putative glucose-methanol-choline oxidoreductase required for the induction of asexual development inAspergillus nidulans.PLoS ONE,7, e40292.
Fu J J, Cheng Y B, Linghu J J, Yang X H, Kang L, Zhang Z X, Zhang J, He C, Du X M, Peng Z Y, Wang B, Zhai L H,Dai C M, Xu J B, Wang W D, Li X R, Zheng J, Chen L,Luo L H, Liu J J,et al. 2013. RNA sequencing reveals the complex regulatory network in the maize kernel.Nature Communications,4, 2832.
Gao J, Zhang Y, Zhang C L, Qi F Y, Li X P, Mu S H, Peng Z H. 2014. Characterization of the floral transcriptome of Moso Bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-Seq analysis.PLoS ONE,9, e98910.
Hao Z F, Li X H, Liu X L, Xie C X, Li M S, Zhang D G, Zhang S H. 2010. Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize.Euphytica,174, 165–177.
Hao Z F, Li X H, Su Z J, Xie C X, Li M S, Liang X L, Weng J F, Zhang D G, Li L, Zhang S H. 2011. A proposed selection criterion for drought resistance across multiple environments in maize.Breeding Science,61, 101–108.
Kakumanu A, Ambavaram M M R, Klumas C, Krishnan A,Batlang U, Myers E, Grene R, Pereira A. 2012. Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq.Plant Physiology,160, 846–867.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T,Yamanishi Y. 2008. KEGG for linking genomes to life and the environment.Nucleic Acids Research,36, 480–484.
Li E, Wang S, Liu Y, Chen J G, Douglas C J. 2011. OVATE FAMILY PROTEIN4 (OFP4) interaction with KNAT7 regulates secondary cell wall formation inArabidopsis thaliana.The Plant Journal,67, 328–341.
Li R Q, Yu C, Li Y R, Lam T W, Yiu S M, Kristiansen K, Wang J. 2009. SOAP2: An improved ultrafast tool for short read alignment.Bioinformatics,25, 1966–1967.
Liu C L, Hao Z F, Zhang D G, Xie C X, Li M S, Zhang X C,Yong H J, Zhang S H, Weng J F, Li X H. 2015. Genetic properties of 240 maize inbred lines and identity-by-descent segments revealed by high-density SNP markers.Molecular Breeding,35, 1–12.
Livak K J, Schmittgen T D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method.Methods,25, 402–408.
Lokesh, kiranmai, Pandurangaiah M, Babu S, Kumar N,Sudhakar C. 2013. Role of plant fatty acid elongase (3 keto acyl-CoA Synthase) gene in cuticular wax biosynthesis.Journal of Agriculture and Allied Sciences,2, 35–42.
Matsuno M, Compagnon V, Schoch G A, Schmitt M, Debayle D, Bassard J E, Pollet B, Hehn A, Heintz D, Ullmann P, Lapierre C, Bernier F, Ehlting J, Werck-Reichhart D.2009. Evolution of a novel phenolic pathway for pollen development.Science,325, 1688–1692.
Mortazavi A, Williams B A, Mccue K, Schaeffer L, Wold B. 2008.Mapping and quantifying mammalian transcriptomes by RNA-Seq.Nature Methods,5, 621–628.
Nakashima K, Yamaguchi-Shinozaki K, Shinozaki K. 2014. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat.Frontiers in Plant Science,5,621–627.
Nariya H, Inouye S. 2002. Activation of 6-phosphofructokinaseviaphosphorylation by Pkn4, a protein Ser/Thr kinase ofMyxococcus xanthus.Molecular Microbiology,46,1353–1366.
Okoniewski M J, Miller C J. 2006. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations.BMC Bioinformatics,7, 276–279.
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K, Tran L S. 2014. ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity.The New Phytologist,202, 35–49.
Perez-Diaz J, Wu T M, Perez-Diaz R, Ruiz-Lara S, Hong C Y,Casaretto J A. 2014. Organ- and stress-specific expression of theASRgenes in rice.Plant Cell Reports,33, 61–73.
Saini H S. 1997. Effects of water stress on male gametophyte development in plants.Sex Plant Reprot,10, 67–73.
Shen C X, Li D, He R H, Fang Z, Xia Y M, Gao J, Shen H, Cao M L. 2014. Comparative transcriptome analysis of RNA-seq data for cold-tolerant and cold-sensitive rice genotypes under cold stress.Journal of Plant Biology,57,337–348.
Shinozaki K, Yamaguchi-Shinozaki K, Seki M. 2003. Regulatory network of gene expression in the drought and cold stress responses.Current Opinion in Plant Biology,6, 410–417.
Tange T, Nott A, Moore M J. 2004. The ever-increasing complexities of the exon junction complex.Current Opinion in Cell Biology,16, 279–284.
Todaka D, Shinozaki K, Yamaguchi-Shinozaki K. 2015. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants.Frontiers in Plant Science,6, 84.
Wang W J, Liu X W, Gai X S, Ren J J, Liu X F, Cai Y L, Wang Q, Ren H Z. 2015.Cucumis sativusL.WAX2plays a pivotal role in wax biosynthesis, influencing pollen fertility and plant biotic and abiotic stress responses.Plant and Cell Physiology,56, 1339–1354.
Wang Z, Gerstein M, Snyder M. 2009. RNA-seq: A revolutionary tool for transcriptomics.Nature Reviews Genetics,10,57–63.
Xiong L, Schumaker K S, Zhu J K. 2002. Cell signaling during cold, drought, and salt stress.The Plant Cell,14, 165–183.
Xu H, Gao Y, Wang J B. 2012. Transcriptomic analysis of rice(Oryza sativa) developing embryos using the RNA-Seq technique.PLoS ONE,7, e30646.
Yin F Q, Qin C, Gao J, Liu M, Luo X R, Zhang W Y, Liu H J, Liao X H, Shen Y U, Mao L K, Zhang Z M, Lin H J, Lubberstedt T,Pan G T. 2015. Genome-wide identification and analysis of drought-responsive genes and microRNAs in tobacco.Inter national Journal of Molecular Sciences,16, 5714–5740.
Zhuang Y, Ren G J, Yue G D, Li Z X, Qu X, Hou G H, Zhu Y, Zhang J R. 2007. Effects of water-deficit stress on the transcriptomes of developing immature ear and tassel in maize.Plant Cell Reports,26, 2137–2147.
杂志排行

Journal of Integrative Agriculture的其它文章
- Improve access to the EU market by identifying French consumer preference for fresh fruit from China
- Management and prevention of mastitis: A multifactorial approach with a focus on milking, bedding and data-management
- Elimination of ceftiofur hydrochloride residue in postpartum cows’milk after intramammary infusing at dry-off
- Evaluation of a new qPCR test to identify the organisms causing high total bacterial count in bulk tank milk
- Prevalence and characteristics of extended spectrum β-lactamaseproducing Escherichia coli from bovine mastitis cases in China
- Evolutionary analysis of plant jacalin-related lectins (JRLs) family and expression of rice JRLs in response to Magnaporthe oryzae