洪泽湖湿地不同植物作用下沉积物细菌群落结构
2018-06-06伍贤军韩建刚李萍萍
伍贤军 ,杨 红 ,程 睿 ,盛 怡 ,韩建刚 ,李萍萍 *
(1.南京林业大学生物与环境学院,南京 210037;2.南京林业大学江苏省南方现代林业协同创新中心,南京 210037)
自然湿地是具有独特结构与复杂功能的生态系统,在维持地球生态平衡,保护生物多样性,促进元素生物地球化学循环等方面发挥着重要作用。江苏淮安洪泽湖湿地是我国第四大淡水湖泊,自然环境条件独特,湿地生物资源十分丰富。主要的植物种类有芦苇、荷花、水花生、茭草、浮萍、金鱼藻、菹草等[1],尤以挺水植物芦苇、荷花和茭草分布较广,植物群落结构比较简单。研究表明,植物对湖泊沉积物有机质的含量有显著影响,水生植物的沉积是湖泊沉积物有机碳和氮的主要来源[2]。同时,沉积物中的有机碳和氮显著影响微生物生物量、群落结构及多样性[3-5]。不同植物类型由于生物量、生长发育期和凋落物性质等不同,对沉积物有机质的贡献存在差异,对自然界的碳氮循环也造成不同的影响。在不同植物的影响下,沉积物有机质含量的差异也会带来微生物群落结构的差异,这种差异展现出来的某些功能菌(比如反硝化细菌)则加速了碳氮循环的进程。目前虽然对植物和有机质,微生物和环境因子,植物和微生物之间的相互关系有较多的研究[6-7],但是对植物-有机质-微生物三者之间的相互关联缺乏认识,对不同植物影响沉积物微生物群落结构的机制尚不明确。另外,微生物在湿地沉积物微环境中扮演着极为重要的角色,如植物残体的分解、污染物的去除、营养元素的物质循环等。对微生物群落结构的解析将为揭开湿地生物反应器这个“黑匣子”的运行机制提供数据基础,为了解湿地环境中微生物与植物,微生物与环境因子,以及微生物之间的相互关系提供依据。
解析湿地沉积物微生物群落结构的方法多种多样[8],如末端限制性片段长度多态性(T-RFLP)技术[9],基于16srRNA和功能基因的RT-qPCR方法[10],磷脂脂肪酸(PLFA)技术[11]以及PCR-DGGE的方法[12]等,这些方法存在检测到的微生物种类不多,微生物分类水平较低等缺陷。近年来,高通量测序技术得到了快速发展,已经逐步应用到生物、医学、农业、食品、环境等各个研究领域[13-17]。基于生物化学手段的PLFA技术以及传统的Q-PCR、TGGE、DGGE等分子生物学方法尽管在环境微生物群落结构研究方面仍然十分有效,但利用高通量测序技术对湿地微生物群落结构进行更为精细的分析十分必要。
本论文研究目的是利用高通量测序技术对洪泽湖湿地西南部河湖交汇区芦苇、茭草和荷花3种不同植物分区沉积物细菌群落结构进行分析,揭示其细菌群落组成和丰度,探讨沉积物细菌群落特征、植物种类、有机质含量三者之间的相互关联,为理解洪泽湖湿地碳氮的生物地球化学循环,挖掘湿地微生物功能奠定基础,为污染物的植物、微生物修复技术提供参考。
1 材料和方法
1.1 样品的采集
洪泽湖湿地处于淮河中下游(33°6′~33°40′N,118°10′~118°52′E),主要分布在环湖地区。其西南部老子山镇是淮河与洪泽湖的交汇地区,具有较为复杂的地理环境因素、特殊的生物资源以及微生物种类。本文以洪泽湖西南部的河湖交汇区(33°12′N,118°33′E)为研究区域。如图1所示,该研究区分布着芦苇、茭草和荷花3种植物群落。3种植物区间隔明显,没有其他的植物类型,面积较大,植物长势较为一致,水流较慢。利用5点取样法[18],于2015年5月在荷花区、茭草区和芦苇区分别采取沉积物样品,采样深度为沉积物表层0~5 cm,将每个区的5个样品混合成一个样品。混合样品置于无菌的50 mL离心管中,并被命名为S1、S2和S3。3份沉积物样品当天带回实验室,-20℃保存。
1.2 理化性质的测定
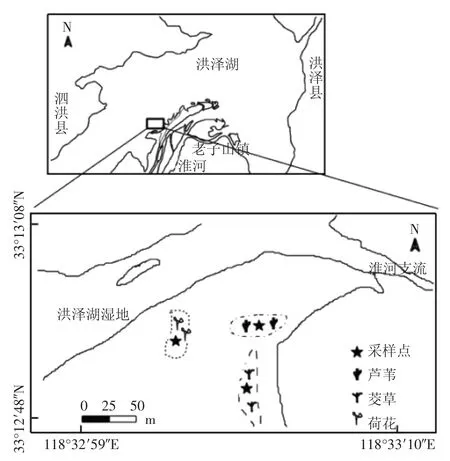
图1 采样点示意图Figure 1 Map of sampling sites
沉积物样品在4℃下解冻。采用鲁如坤[19]描述的方法对样品进行预处理。总有机碳(Total organic carbon,TOC)采用重铬酸钾容量法-外加热法(F-HZ-DZ-TR-0046);总氮(Total nitrogen,TN)采用凯氏定氮法(GB 7848—1987);硝酸盐氮(Nitrate nitrogen,NO3-N)采用HACH分光光度计法8507(HACH DR/2400);氨氮(Ammonia nitrogen,NH3-N)采用 HACH分光光度计法8038(HACH DR/2400);总磷(Total phosphorus,TP)采用碱熔-钼锑抗分光光度法(HJ 632—2011)测定。样品的理化性质指标进行2次重复测定(取2次测定结果平均值)。运用SPSSStatistics 17.0统计软件进行数据分析。
1.3 基因组DNA的提取
采用天根生化科技(北京)有限公司的土壤微生物基因组DNA提取试剂盒(DP336)的方法,称量0.5 g沉积物样品,按试剂盒的实验步骤提取沉积物微生物的总DNA。获得的总DNA样品纯度和质量通过0.8%的琼脂糖凝胶电泳检测。DNA浓度通过Nano Drop ND-1000微型分光光度计测定。
1.4 PCR扩增及高通量测序
为了构建细菌群落的基因文库进行高通量测序,选择细菌16SrRNA的V4-V5可变区作为测序的目标序列。利用通用引物515F(5′-GTGCCAGCMGCCGCGG-3′)和 907R(5′-CCGTCAATTCMTTTRAGTTT-3′),以提取到的总DNA样品为模板PCR扩增。PCR反应体系为 50 μL,包含有 0.2 μmol·L-1的引物,10 ng的DNA 模板,0.25 mmol·L-1dNTPs,1×PCR 反应缓冲液,2U的快速PfuDNA聚合酶(天根公司,北京)。PCR扩增使用的仪器为ABI GeneAmp 9700(USA)。使用的反应条件为:95℃预变性2 min;95℃变性30 s,55℃退火 30 s,72℃延伸 45 s,30个循环;72℃延伸 10 min。PCR产物通过2%的琼脂糖凝胶电泳检测,EB染色后,通过琼脂糖凝胶回收试剂盒(天根公司,北京)对目标条带进行纯化。对纯化的PCR产物进行浓度的测定,以达到高通量测序的要求。将PCR产物样品送至上海美吉生物医药科技有限公司,在Ilumina-Miseq平台上进行高通量测序。
1.5 测序数据处理
测序得到的序列通过以下方法利用Trimmomatic软件进行质控过滤:(1)过滤read尾部质量值20以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,过滤质控后50 bp以下的read;(2)根据 PE reads之间的overlap关系,将成对reads拼接(merge)成一条序列,最小overlap长度为10 bp;(3)拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列。根据序列首尾两端的barcode和引物序列区分样品得到有效序列,并校正序列方向。利用Usearch(vsesion 7.1)软件按照97%相似性对非重复序列进行OTU聚类,在聚类过程中去除嵌合体,得到OTU的代表序列,将所有优化序列map至OTU代表序列,选出与OTU代表序列相似性在97%以上的序列。采用RDPclassifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,并在门和属的水平上统计每个样品的群落组成。选用相似水平为97%的OTU样本,统计多个样本中所共有和独有的OTU数目表,绘制OTU分布韦恩图[20]。对复杂数据降维,运用方差分解,将多组数据的差异反映在二维坐标图上,绘制PCA图[21]。用颜色变化来反映二维矩阵中的数据信息,并将数据进行物种或样本间丰度相似性聚类,绘制群落的结构热图[22]。
2 结果分析
2.1 样品理化指标
为了解研究区沉积物的生理生化条件,我们分析了沉积物样品S1、S2和S3的某些理化指标。如表1所示,S1 的总有机碳(TOC,0.66 g·kg-1)、总氮(TN,0.08 g·kg-1)、氨氮(NH3-N,5.53 mg·kg-1)和硝酸盐氮(NO3-N,1.47 mg·kg-1)含量都明显低于 S2 及S3,特别是总氮(TN)含量只有 S2(0.44 g·kg-1)的 1/5 左右。结合ANVOA方差分析,如表2所示,S1、S2和S3的碳氮含量差异显著(P<0.05),其中S1和S2的差异最大,S2和S3的差异次之,S2和S3的差异最小。3个样品总磷(TP)含量无显著性差异(P>0.05)。表明在同一片水域受外来影响相同的情况下,植物种类可能影响到沉积物有机质的含量,这些有机质是植物本身凋落或死亡的残体,对沉积物中的氮碳含量有着重要的影响。
2.2 测序结果的质量分析
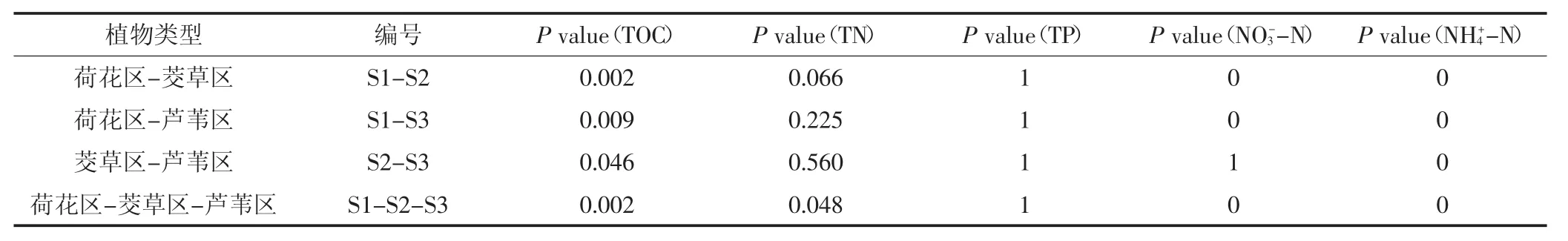
沉积物样品S1、S2和S3分别得到19 522、17 697和14 785条有效序列,获得1494、1503、1600个OTU。通过输入序列数目(小于总的样本序列条数)与OTU个数产出间的相互关系,绘制稀疏曲线。根据各样本的测序量在不同测序深度OTU个数不再有升高的趋势来表明测序数据是否完全覆盖微生物群落全部的多样性。由图2可知,所有样品曲线均趋于平缓,这表明实际测序量完全覆盖了群落物种的组成,可以真实反映群落各物种间的相对比例关系。
2.3 不同植物作用下沉积物微生物多样性
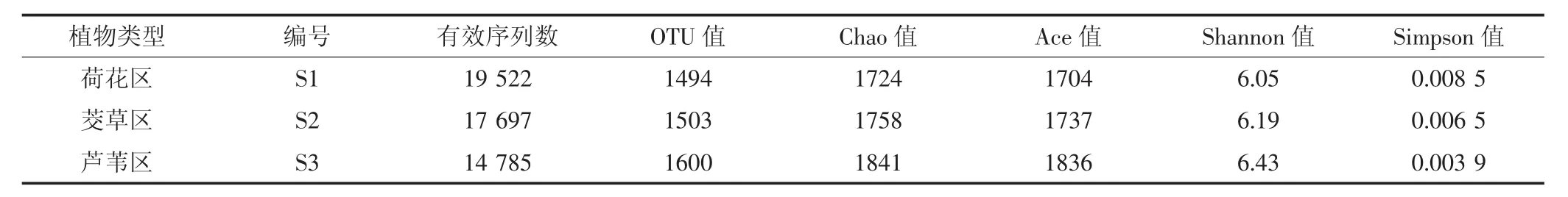
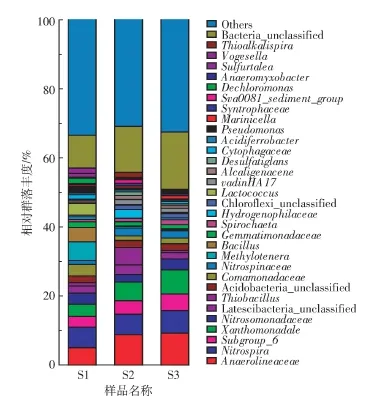
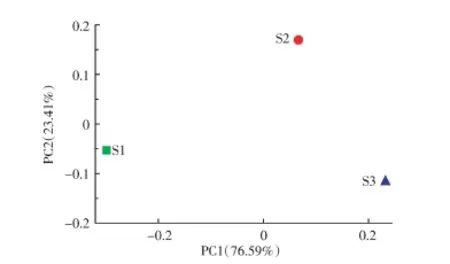
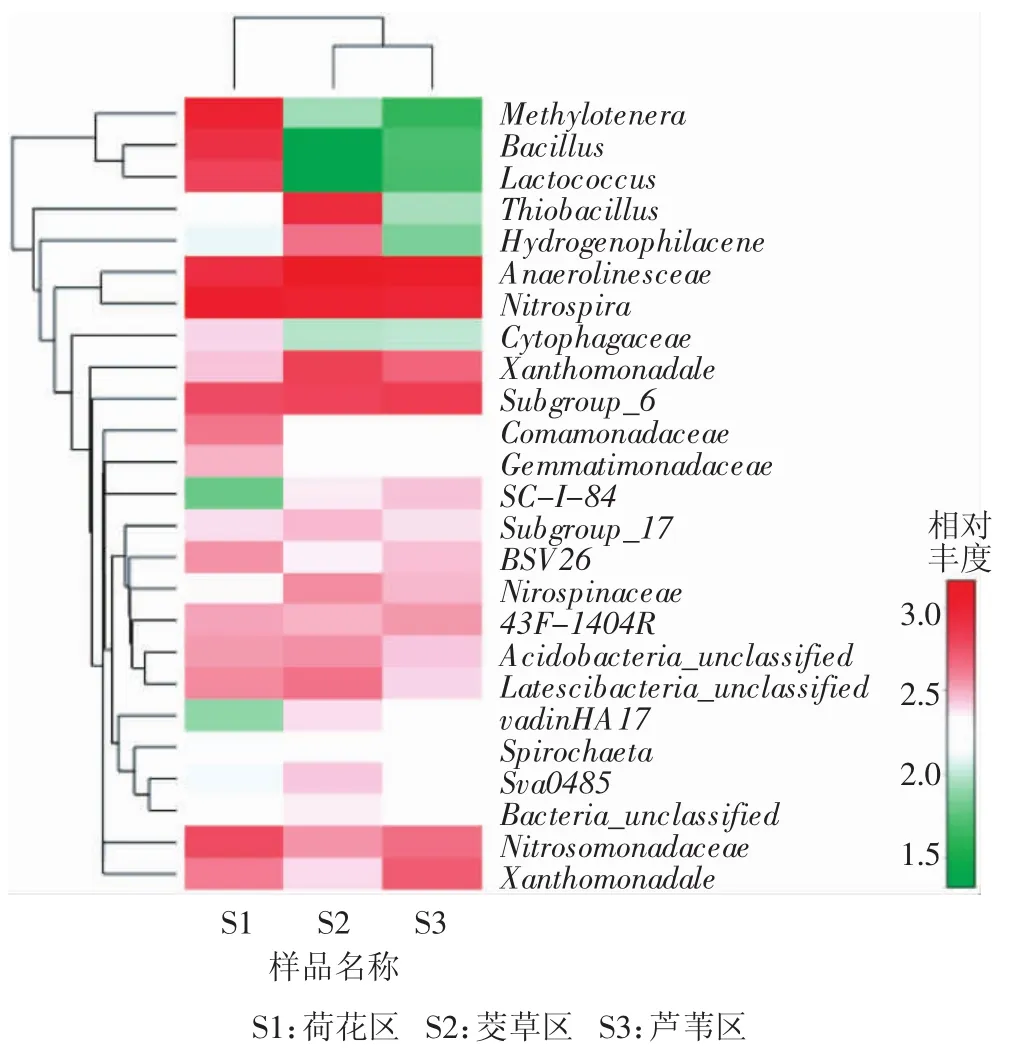
对单样品的α多样性分析可以反映微生物的丰度及多样性,包括利用Chao值、Ace值、Shannon值和Simpson值等一系列统计学分析指数对微生物多样性的大小进行估算。Chao值、Ace值和Shannon值越大,Simpson值越小,说明样品中物种越丰富。如表3所示,Chao值和Ace值芦苇区S3最高,茭草区S2其次,荷花区S1最低,从Shannon指数和Simpson指数来看,微生物物种丰度也表现为S1 表1 洪泽湖湿地沉积物的理化特征Table 1 Physicochemical properties of sedimentsfrom Hongze Lake wetland 表2 沉积物理化特征差异显著性比较Table 2 Comparison of differencesignificant of physicochemical propertiesof sediments 将3种沉积物样品S1、S2和S3获得的OTU进行注释,统计在门类别上的物种组成,如图3所示。共有14个门的物种占所在样品的比例在1%以上。3种沉积物样品都以变形菌门(Proteobacteria)为优势菌群,占所在样品的比例均接近50%。其他的主要菌群为绿弯菌门(Chloroflexi,占 7.2%~14.7%)、酸杆菌门(Acidobacteria,占9.47%~12.18%)、硝化螺旋菌门(Nitrospirae,占 6.14%~7.15%),拟杆菌门(Bacteroidetes,占4.14%~4.34%),不同的植物类型之间以上菌群占所在样品的比例均表现出一定的差异。而更大的差异来自厚壁菌门(Firmicutes)。荷花区沉积物样品S1的厚壁菌门(Firmicutes)占所在样品的比例高达10.48%,而茭草区S2、芦苇区S3的厚壁菌门(Firmicutes)非常少(占1%左右)。这表明荷花区沉积物的微生物群落组成与芦苇区、茭草区比较具有明显的差异,而厚壁菌门(Firmicutes)丰度高是其主要的特征。3种沉积物样品中还含有一定比例的绿菌门(Chorobi,占 2.38%~2.87%)、浮霉菌门(Planctomycetes,占 1.79%~2.31%)、放线菌门(Actinobacteria,占1.15%~2.18%),各样品所占比例相差不大。另外,沉积物中也有Latescibacteria、芽单胞菌门(Gemmatimonadetes)、螺旋菌门(Spirochaetae)、Aminicenadetes的细菌,但丰度很低,有些占所在样品的比例不到1%。 图2 洪泽湖湿地沉积物稀释性曲线Figure2 Rarefaction curvefromsedimentsof Hongze Lakewetland 3种沉积物样品中微生物群落在属分类水平上的分布情况如图4所示。其中硝化螺旋菌属(Nitrospira)在3种沉积物样品中都占有较大优势(6.14%~7.14%)。其在沉积物环境中起主要的硝化作用,可把亚硝酸盐转化为硝酸盐[23],最近发现某些硝化螺旋菌也能展现完全的硝化能力,直接把氨盐氧化成硝酸盐[24]。亚硝酸单胞菌(Nitrosomonadaceae)也占有较高的比例(2.36%~3.45%),负责把氨盐氧化成亚硝酸盐[25]。这些细菌在3种沉积物样品中的丰度未表现出明显的差异,植物类型对其丰度影响不大。S2和S3还含有相当比例的Nitrospinaceae,分别占2.25%和2.36%,而荷花区S1丰度较低,占1.24%,它们在环境中也具有一定的硝化作用。3种沉积物样品含有丰富的厌氧绳菌科(Anaerolineaceae),它们在S2和S3中的比例高达8.85%和9.1%,而在S1中也偏低,占4.86%。厌氧绳菌科(Anaerolineaceae)是严格的厌氧菌,属于绿弯菌门(Chloroflexi),在产甲烷烷烃降解中扮演重要的角色[26]。比较而言,与甲醇代谢有关的嗜甲基菌(Methylotenera)含量在S1中的丰度较高,所占比例达到5.77%,而在S2和S3中仅占0.5%和0.3%。同样的情况出现在厚壁菌门(Firmicutes)的芽孢杆菌(Bacillus)和乳球菌(Lactococcus)上,它们在 S1中含量为4.47%和3.61%,而在S2和S3中仅为0.1%和0.3%,这与厚壁菌门(Firmicutes)在S1中的含量远高于S2和S3的结果一致。除此之外,S1含有相当比例的假单胞菌(Pseudomonas,1.87%),而 S2和 S3几乎没有。另外,沉积物样品S1、S2和S3中还含有3.88%、5.74%和7.63%的黄色单胞菌目(Xanthomonadale)的细菌。其他的微生物主要还有丛毛单胞菌科(Comamonadaceae)、嗜氢菌科(Hydrogenophilaceae)、未分类的 Latescibacteria、福格斯氏菌(Vogesella)、噬纤维菌科(Cytophagaceae)等,它们在某些样品中占的比例也在1%以上。以上结果表明,荷花区沉积物S1与芦苇或茭草区沉积物S2或S3在细菌群落组成上存在较大差异,其嗜甲基菌(Methylotenera)、芽孢杆菌(Bacillus)、乳球菌(Lactococcus)和假单胞菌(Pseudomonas)丰度高出芦苇区或茭草区的10倍以上。 表3 洪泽湖湿地沉积物细菌多样性指数Table3 The bacterial diversity index fromsediment samples of the Hongze Lakewetland 图3 洪泽湖湿地沉积物微生物群落结构组成分布(门水平)Figure3 Bacterial community composition at phylumlevel in sediment samplesof the Hongze Lake wetland 图4 洪泽湖湿地沉积物微生物群落结构组成分布(属水平)Figure 4 Bacterial community composition at genus level in sediment samplesof the Hongze Lake wetland 维恩图是用封闭曲线直观地表示集合及其关系的图形,通过维恩图可以展示3个沉积物样品之间OTU的相似性及分布情况。如图5所示,S1和S2共有 OTU为 890,S2和 S3共有 OTU 为 952,S1和 S3共有OTU为959,3个样品共有的OTU数目则为836,主要是硝化细菌。S1独有的OTU为73,S2独有的OTU为23,S3独有的OTU为16,表明S1具有较多独特的微生物种类,主要是嗜甲基菌(Methylotenera)、假单胞菌(Pseudomonas)以及厚壁菌门(Firmi cutes)的细菌。基于OTU的主成分分析表明样品菌落之间的差异,如图6所示,在横坐标方向,物种累计方差贡献率达到76.59%,沉积物样品S2、S3相距较近,而与S1相距较远,表明S1与S2、S3都具有较大差异,即荷花区沉积物与芦苇区和茭草区比较,细菌群落结构具有较大差异。 微生物结构热图可以反映样品之间的物种组成和差异,也可以对物种和样本进行聚类分析。如图7所示,在属的分类水平上,对总丰度前25的物种进行热图分析表明,S1和S2、S3在物种组成上具有较大差异,这种差异主要表现在S1含有较多的嗜甲基菌(Methylotenera)以及厚壁菌门的芽孢杆菌(Bacillus)和乳球菌(Lactococcus),而S2和S3这些菌群则很少。样品聚类分析也显示S2和S3群落结构具有比较高的相似性,它们与S1存在一定的差异。物种聚类分析表明:芽孢杆菌(Bacillus)和乳球菌(Lactococcus)、厌氧绳菌科(Anaerolineaceae)和硝化螺旋菌属(Nitrospira)、未分类的Latescibacteria和Acidobacteria群落分布较为类似,它们在沉积物中的功能可能具有一定的相关性。 图5 洪泽湖湿地沉积物中微生物OTU分布Venn图Figure5 Venn diagramof bacteriafromsedimentsof the Hongze Lakewetland 图6 洪泽湖湿地沉积物微生物群落组成的UniFrac PCA分析Figure 6 The weighted UniFrac PCA analysisof thebacterial community fromsedimentsof the Hongze Lakewetland 图7 沉积物微生物群落组成热图(属水平)Figure 7 Heat map showing bacterial composition fromsediments on genus level 湿地植物对湖泊沉积物中有机碳和氮的含量有重要的影响。研究表明,湿地植物的存在显著地增加了沉积物中碳氮的含量,湖泊内部水生植物的残体沉积是沉积物有机质的主要来源,不同的植物类型与沉积物有机质的含量密切相关[27-28]。在同一片区域,可以认为外源有机碳和氮影响一致,仅植物种类影响沉积物的碳氮含量。在本研究中,荷花区沉积物的总有机碳、全氮、氨氮和硝酸盐氮比芦苇区、茭草区要低得多,存在极显著的差异。这可能与它们不同的生物量有关。在洪泽湖湿地,芦苇的生物量大于荷花的生物量[29],芦苇凋落物和残体沉积的量应大于荷花的量,其凋落物的性质也可能具有一定的差异,而凋落物和残体沉积是有机质的主要来源[30]。茭草和芦苇的碳氮含量没有显著差异,推测它们具有相近的生物量,但还需要更多的实验证实。环境因子与沉积物微生物群落结构密切相关,这些因子包括有机质、重金属、酸碱度、含水率以及氧化还原电位(ORP)等,碳氮含量是影响微生物群落结构主要的环境因子[31]。在本研究中,碳氮含量较低的荷花区沉积物细菌群落结构与碳氮含量较高的芦苇区或茭草区具有明显差异,这表明水生植物通过改变沉积物有机质的含量影响其微生物群落结构,而不同的植物产生有机质含量的差异是造成沉积物微生物群落结构差异的重要原因。具体来看,碳氮含量较低的荷花区沉积物含有丰富的厚壁菌门(Firmicutes),碳氮含量较高的芦苇区和茭草区则都很少。这种极为显著的差异也表明沉积物有机质和厚壁菌门(Firmicutes)的细菌具有更为密切的关联。从分类学水平更高的属上看,厚壁菌门(Firmicutes)的芽孢杆菌和乳球菌在荷花区沉积物中具有相对高的丰度。对于湿地沉积物中有机质和这类细菌关系的机制尚不清楚。荷花区沉积物也还含有较高的假单胞菌(Pseudomonas)、嗜甲基菌(Methylotenera),这些菌株在芦苇区、茭草区沉积物中含量则极少。有机质对微生物结构多样性的影响必然引起微生物功能多样性的变化。研究表明,芽孢杆菌(Bacillus)具有较强的好氧反硝化能力,在反硝化细菌群体中占据了较大的比重[32]。假单胞菌(Pseudomonas)也是常见的反硝化细菌,很多的反硝化菌被鉴定为荧光假单胞菌、施氏假单胞菌、恶臭假单胞菌、硝基还原假单胞菌和嗜麦芽假单胞菌等[33]。嗜甲基菌(Methylotenera)是一种最新发现的与甲醇代谢有关的变形菌,同时具有反硝化能力[34]。因此,可以推测,荷花区沉积物存在较强的好氧反硝化过程,具有更强的反硝化潜力,有利于富营养化湖泊中氮素的去除,加速氮元素的生物地球化学循环,但仍需更多的实验证实。 相对于荷花,芦苇和茭草沉积物相对丰富的有机质含量必然会引起好氧环境的减弱和厌氧环境的增强,这可能是好氧反硝化在荷花区较强的原因。这种好氧环境和厌氧环境的此消彼长也能很好地解释为何芦苇和茭草沉积物均含有相当高比例的厌氧蝇菌科(Anaerolineaceae)的细菌,而荷花区比例较低。 (1)在3种植物芦苇、茭草和荷花区的沉积物中,均以变形菌门(Proteobacteria)为优势菌群,相对丰度比例相似,都接近50%;硝化螺旋菌属(Nitrospira)和亚硝化单胞菌属(Nitrosomonadaceae)也占有较大的比例,且相对丰度相当。沉积物硝化过程活跃,植物类型对其没有影响。 (2)在相同的湿地环境中,荷花比芦苇或茭草具有更低的碳氮水平,更低的细菌丰度及多样性。与芦苇和茭草相比,荷花沉积物细菌群落组成呈现出较大差异,出现丰富的厚壁菌门(Firmicutes)的细菌,如芽孢杆菌(Bacillus)和乳球菌(Lactococcus)等。植物通过改变沉积物碳氮含量影响其细菌群落结构,而不同的植物作用下沉积物碳氮含量的差异是造成其细菌群落结构差异的重要原因。 (3)与芦苇和茭草相比,荷花区沉积物不仅有相对丰富的芽孢杆菌(Bacillus)和乳球菌(Lactococcus),还有高比例的假单胞菌(Pseudomonas)和嗜甲基菌(Methylotenera)等,它们都具有较强的好氧反硝化功能,这暗示荷花区沉积物有更强的反硝化潜力,可为湖泊富营养化的植物修复提供借鉴和参考。 [1]潘宝宝,张金池,冯开宇,等.洪泽湖典型水生植物群落碳储量[J].湿地科学,2014,12(4):471-476.PANBao-bao,ZHANGJin-chi,FENGKai-yu,et al.Carbon storage of typical aquatic plant communitiesin Hungtse Lake[J].Wetland Science,2014,12(4):471-476. [2]倪兆奎,李跃进,王圣瑞,等.太湖沉积物有机碳与氮的来源[J].生态学报,2011,31(16):4661-4670.NI Zhao-kui,LI Yue-jin,WANG Sheng-rui,et al.The sources of organic carbon and nitrogen in sediment of Taihu Lake[J].Acta Ecologica Sinica,2011,31(16):4661-4670. [3]杜瑞芳,李靖宇,赵 吉.乌梁素海湖滨湿地细菌群落结构多样性[J].微生物学报,2014,54(10):1116-1128.DU Rui-fang,LI Jing-yu,ZHAO Ji.Bacterial diversity in littoral wetland of Wuliangsuhai Lake[J].Acta Microbiologica Sinica,2014,54(10):1116-1128. [4]冯 峰,王 辉,方 涛,等.东湖沉积物中微生物量与碳、氮、磷的相关性[J].中国环境科学,2006,26(3):342-345.FENGFeng,WANGHui,FANGTao,et al.The correlation between microbial biomass and carbon,nitrogen,phosphorus in the sediments of Lake Donghu[J].China Environmental Science,2006,26(3):342-345. [5]王 娜,徐德琳,郭 璇,等.太湖沉积物微生物生物量及其与碳、氮、磷的相关性[J].应用生态学报,2012,23(7):1921-1926.WANG Na,XU De-lin,GUO Xuan,et al.Microbial biomass and its correlations with carbon,nitrogen,and phosphorus in the sediments of Taihu Lake[J].Chin JAppl Ecol,2012,23(7):1921-1926. [6]陆开宏,胡智勇,梁晶晶,等.富营养水体中2种水生植物的根际微生物群落特征[J].中国环境科学,2010,30(11):1508-1515.LUKai-hong,HU Zhi-yong,LIANGJing-jing,et al.Characteristics of rhizosphere microbial community structure of two aquatic plants in eutrophic waters[J].China Environmental Science,2010,30(11):1508-1515. [7]张亚朋,章婷曦,王国祥.苦草(Vallisneria natans)对沉积物微生物群落结构的影响[J].湖泊科学,2015(3):445-450.ZHANG Ya-peng,ZHANG Ting-xi,WANG Guo-xiang.Influence of Vallisneria natans on microbial community in sediments[J].Journal of Lake Sciences,2015,27(3):445-450. [8]李甜甜,胡 泓,王金爽,等.湿地土壤微生物群落结构与多样性分析方法研究进展[J].土壤通报,2016(3):758-762.LITian-tian,HU Hong,WANGJin-shuang,et al.Progress in research methodsof soil microbial structureand diversity in wetlands[J].Chinese Journal of Soil Science,2016(3):758-762. [9]Peralta A L,Matthews JW,Kent A D.Microbial community structure and denitrification in awetland mitigation bank[J].Applied&Environmental Microbiology,2010,76(13):4207-4215. [10]Stoeva M K,Aris-Brosou S,Chételat J,et al.Microbial community structure in lakeand wetland sedimentsfromahigh Arctic polar desert revealed by targeted transcriptomics[J].PLoS One,2014,9(3):e89531-e89531. [11]张丁予,章婷曦,董丹萍,等.沉水植物对沉积物微生物群落结构影响:以洪泽湖湿地为例[J].环境科学,2016,37(5):1734-1741.ZHANGDing-yu,ZHANGTing-xi,DONGDan-ping,et al.Influence of submerged plants on microbial community structure in sediment of Hongze Lake[J].Environmental Science,2016,37(5):1734-1741. [12]刘绍雄,王明月,王 娟,等.基于PCR-DGGE技术的剑湖湿地湖滨区土壤微生物群落结构多样性分析[J].农业环境科学学报,2013,32(7):1405-1412.LIUShao-xiong,WANGMing-yue,WANGJuan,et al.Analyzing soil microbial community structure diversity from Jianhu wetland lakeside zone using PCR-DGGE technique[J].Journal of Agro-Environment Science,2013,32(7):1405-1412. [13]Schuster SC.Next-generation sequencing transforms today′s biology[J].Nature Methods,2007,5(1):16-18. [14]Dorn C,Grunert M,Sperling SR.Application of high-throughput sequencingfor studyinggenomic variationsin congenital heart disease[J].Briefingsin Functional Genomics,2014,13(1):51-65. [15]Li X R,Liu X F,Zhang H Y,et al.Microbial community diversity in douchi from Yunnan Province using high-throughput sequencing technology[J].Modern Food Science&Technology,2014,30(12):61-67. [16]Shapter F M,Cross M,Ablett G,et al.High-throughput sequencing and mutagenesistoacceleratethedomestication of Microlaenastipoides asanew food crop[J].Plos One,2013,8(12):e82641. [17]庄林杰,夏 超,田 晴,等.高通量测序技术研究典型湖泊岸边陆向深层土壤中厌氧氨氧化细菌的群落结构[J].环境科学学报,2017,37(1):261-271.ZHUANGLin-jie,XIA Chao,TIAN Qing,et al.Community structure of anaerobic ammoniaoxidation bacteria in subsurface soil from riparian zonetoland in Baiyangdian Lakeusinghigh-throughput sequencing technology[J].Acta Scientiae Circumstantiae,2017,37((1):261-271. [18]张 昕,简桂良,林 玲,等.土壤中落叶型棉花黄萎病菌的分子检测方法[J].江苏农业学报,2011,27(5):990-995.ZHANGXin,JIANGui-liang,LINLing,et al.Detection of defoliating pathotype of Verticillium dahliae in soil by nested-PCR[J].Jiangsu Journal of Agricultural Sciences,2011,27(5):990-995. [19]鲁如坤.土壤农业化学分析方法[M].北京:中国农业科技出版社,2000:147-211.LU Ru-kun.Soil agrochemistry analysis protocoes[M].Beijing:China Agriculture Science Press,2000:147-211. [20]Fouts DE,Szpakowski S,Purushe J,et al.Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J].PLoS One,2012,7(11):e48289. [21]Oberauner L,Zachow C,Lackner S,et al.The ignored diversity:complex bacterial communities in intensive care units revealed by 16Spyrosequencing[J].Scientific Reports,2013,3(3):1413. [22]Jami E,Israel A,Kotser A,et al.Exploring the bovine rumen bacterial community frombirth toadulthood[J].Isme Journal,2013,7(6):1069. [23]Watson SW,Bock E,Valois FW,et al.Nitrospiramarina,gen.nov.sp.nov.:A chemolithotrophic nitrite-oxidizing bacterium[J].Archives of Microbiology,1986,144(1):1-7. [24]Daims H,Lebedeva E V,Pjevac P,et al.Complete nitrification by Nitrospirabacteria[J].Nature,2015,528(7583):504. [25]Lynch A.Bergey′s manual of systematic bacteriology:Volume two Proteobacteria(Part C)-The Proteobacteria[M].Berlin:Springer,2011:89-100. [26]Liang B,Wang L Y,Mbadinga S M,et al.Anaerolineaceae and Methanosaeta,turned to be the dominant microorganisms in alkanesdependent methanogenic culture after long-termof incubation[J].Amb Express,2015,5(1):37. [27]余 辉,张文斌,卢少勇,等.洪泽湖表层底质营养盐的形态分布特征与评价[J].环境科学,2010,31(4):961-968.YU Hui,ZHANG Wen-bin,LU Shao-yong,et al.Spatial distribution characteristicsof surface sedimentsnutrients in Lake Hongzeand their pollution status evaluation[J].Environmental Science,2010,31(4):961-968. [28]黄玉洁,张 勇,张银龙,等.太湖百渎港湿地植物群落沉积物中碳、氮的空间变化研究[J].环境科学与管理,2015,40(3):140-145.HUANG Yu-jie,ZHANG Yong,ZHANG Ying-Long,et al.Spatial variation of Cand Nin sedimentsof wetland plant community in Baidu Port of Taihu Lake[J].Environmental Science&Management,2015,40(3):140-145. [29]潘宝宝.洪泽湖湿地水生植物群落碳储量研究[D].南京:南京林业大学,2013.PANBao-bao.Research on carbon storageof major aquatic plant communities[D].Nanjing:Nanjing Forestry University,2013. [30]葛绪广,王国祥,李振国,等.凤眼莲凋落物及其残体的沉降[J].湖泊科学,2009,21(5):682-686.GE Xu-guang,WANG Guo-xiang,LI Zhen-guo,et al.The litter and residue of Eichhornia crassipes(Mart.)Solms[J].Journal of Lake Sciences,2009,21(5):682-686. [31]金 笑,寇文伯,于昊天,等.鄱阳湖不同区域沉积物细菌群落结构、功能变化及其与环境因子的关系[J].环境科学研究,2017,30(4):529-536.JING Xiao,KOU Wen-bo,YU Hao-tian,et al.Environmental factors influencing the spatial distribution of sediment bacterial community structure and function in Poyang Lake[J].Research of Environmental Sciences,2017,30(4):529-536. [32]Kim JK,Park K J,Cho K S,et al.Aerobic nitrification-denitrification by heterotrophic Bacillus strains[J].Bioresource Technology,2005,96(17):1897-1906. [33]Ji B,Yang K,Zhu L,et al.Aerobic denitrification:A review of important advancesof thelast 30 years[J].Biotechnology and Bioprocess Engineering,2015,20(4):643-651. [34]Mustakhimov I,Kalyuzhnaya M G,Lidstrom M E,et al.Insights into denitrification in Methylotenera mobilis from denitrification pathway and methanol metabolism mutants[J].Journal of Bacteriology,2013,195(10):2207.
2.4 不同植物作用下沉积物微生物群落组成

2.5 不同植物作用下沉积物微生物群落相似性分析

3 讨论
4 结论
