α-季碳氨基酸的不对称催化合成
2018-06-04周剑
周 剑
(华东师范大学 化学与分子工程学院 绿色化学与化工过程绿色化上海市重点实验室, 上海 200062)
1 α-季碳氨基酸的重要性
氨基酸是指羧酸分子中,一个或多个氢原子被氨基取代后的化合物.顾名思义,α-氨基酸的氨基在羧基的邻位,即α位.所有生命体中的蛋白质主要由20种氨基酸组成,而这些氨基酸均为α-氨基酸.这20种必需氨基酸通常称为蛋白源氨基酸,是生命体中多肽和蛋白质的基本砌块,是生命之源,对生命体的各种代谢和生理活动如识别、转换、交流和调节起着非常重要的作用.其余的氨基酸被称为非蛋白源氨基酸,一般是在蛋白源氨基酸基础上人工设计或修饰而成,在化学、药学和生物等众多领域具有重要的应用.α-季碳氨基酸是一类重要的非蛋白源氨基酸,其特点是与氨基和羧基同时相连的碳上的2个氢原子均被其他基团取代,因此具有很好的结构刚性和多样性.
如图1所示,α-季碳氨基酸广泛存在于天然产物、药物及生物活性分子中[1-2].例如,从放线菌菌株的链霉菌Sioyaensis SA-1758中分离的含有环状α-季碳氨基酸的Altemicidin (1),对肿瘤细胞的生长以及朱砂叶螨具有很强的抑制活性和杀虫活性[3].另外,从菲律宾长滩岛西北部海域的海绵Leiosella中分离得到的天然产物Halipeptin D(2)对人类的结肠癌细胞(HCT-116)具有很强的抑制作用[4];而含三元环结构的α-季碳氨基酸(+)-Coronatine (3)是一种植物毒素,对植物生长与保护起着重要的作用[5-7];从海洋生物中分离出来的Sphingofungin E(4a)和F(4b)对丝氨酸棕榈转移酶具有有效的抑制活性[8-9].除了这些天然产物分子外,非天然的手性α-季碳氨基酸还是许多药物以及药物分子的前体.例如,化合物AG-041R (5)的结构中含有3-取代-3-胺基氧化吲哚这类特殊的α-季碳氨基酸结构单元.它不仅是胃分泌激素CCK-B受体拮抗剂,同时还具有修复软骨缺陷的作用[10];而在鸟氨酸的α位选择性引入一个二氟甲基后形成的依氟鸟氨酸6(Eflornithine),可以有效地治疗面部多毛症或毛发过度生长,以及非洲锥虫病(昏睡病)和脑瘤[11];α-甲基多巴胺7对芳香酸脱羧酶有很强的抑制活性,是常用的治疗高血压的药物[12];辣椒素受体抑制剂 Spirohydatoin I (8)是由阿斯利康公司发现的具有止痛效果的化合物[13].
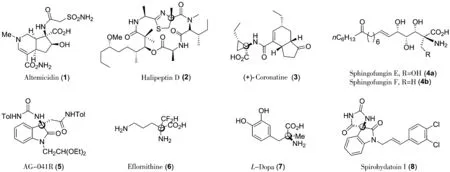
图 1 一些含α-季碳氨基酸结构的天然产物、药物和药物活性分子
由于α-季碳氨基酸的α位不含体积最小的氢原子,因此其结构稳定、刚性较强.例如,在天然或人工设计合成的多肽化合物中引入这类结构受限的刚性结构单元有可能形成稳定的多肽构象,从而使新形成的多肽折叠可控[14-16]或具有抵抗酶降解[17-20]等许多有利的性质.这一特点也为设计生物活性多样的多肽和蛋白质提供了可能性.同时,手性α-季碳氨基酸作为一类廉价的手性源,还有望在材料、催化剂设计以及手性识别等领域展示较好的发展前景.
综上所述,α-季碳氨基酸在天然产物和药物化学等众多研究中具有重要应用价值,多样性合成不同结构和取代基的α-季碳氨基酸有助于研究相关生物活性分子的构效关系,为新药研发提供技术支持.
2 α-季碳氨基酸的不对称催化合成策略
基于α-季碳氨基酸的重要性,发展高效的不对称催化反应来合成手性α-季碳氨基酸受到合成化学家们的广泛关注,并成为当代有机化学领域的研究热点之一.目前,已经发展了一系列不对称催化的合成方法.这些方法可根据反应底物的不同,分为如图2所示的4种合成策略:1) 酮亚胺的不对称Strecker反应;2) α-酮酸酯衍生的酮亚胺的不对称亲核加成反应;3) 不对称胺化反应;4) 基于α-氮原子取代羧酸衍生物的不对称官能团化反应.
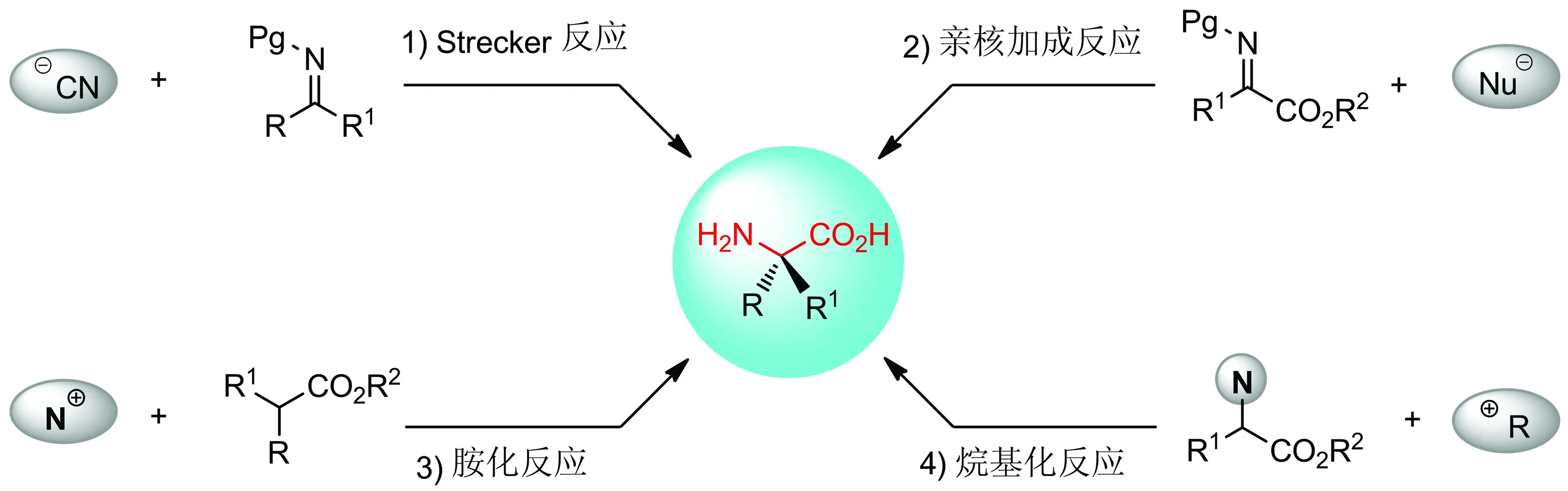
图2α-季碳氨基酸的不对称催化合成策略
Fig.2SyntheticstrategiesofCα-tetrasubstitutedα-aminoacids
我们课题组自2008年底成立以来,一直致力于具有季碳手性中心化合物的不对称催化构建,因此对发展手性α-季碳氨基酸这类优势骨架的合成方法很感兴趣.并利用上述4种策略进行了多方尝试.下面简要介绍我们近十年来取得的研究进展.
3 酮亚胺的不对称Strecker反应
酮亚胺参与的不对称Strecker反应是制备α-季碳氨基酸前体的重要方法.尽管醛亚胺的不对称催化研究已取得了很大进展[21-22],但由于酮亚胺的活性较低、位阻较大,尽管已有一些成功的例子[23-31],但在我们研究工作之初,一些在药物化学中具有重要潜在用途的酮亚胺的Strecker反应还没有人研究.
例如,含3-胺基3-取代氧化吲哚骨架的季碳氨基酸广泛存在于各种药物及生物活性分子中[32-33],但靛红亚胺的不对称Strecker反应尚无人报道.我们利用自主发展的新型手性膦酰胺-叔胺双功能催化剂10实现了TMSCN(三甲基氰硅烷)对N-芳基取代的靛红亚胺9的不对称Strecker反应,以中等产率和对映选择性获得了手性3-胺基氧化吲哚类化合物11(图 3)[34].虽然该反应的产率和对映选择性还有很大提升空间,但这是首例不对称催化的亲核试剂对靛红酮亚胺的加成反应,为不对称合成胺基氧化吲哚开辟了一条新的途径.
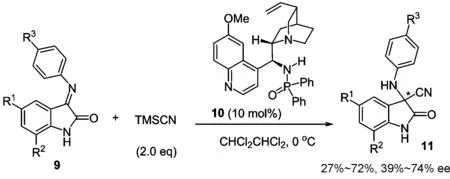
图 3 N-PMP靛红酮亚胺9的不对称Strecker反应
为了进一步提高产率和对映选择性,我们后续研究发现活性更高的N-Boc靛红酮亚胺12,可以在金鸡纳碱衍生的手性双功能硫脲催化剂13a的催化下,很好地与TMSCN发生不对称Strecker反应,以中等到优秀的产率及优秀的对映选择性得到目标化合物14(图 4)[35].对照实验显示,双功能催化剂中叔胺与硫脲的协同作用对该反应的高效和高选择性至关重要,因为当催化剂中硫脲的一个N—H键被甲基保护后所得的催化剂13b给出了明显更低的产率和对映选择性.基于此,我们提出了如图 4所示的双功能协同活化的反应过渡态,催化剂可以有效地以类似于分子内的方式进行组织反应.

图 4 N-Boc靛红酮亚胺12的不对称Strecker反应
由于N-Boc靛红亚胺在分离纯化过程中产率损失较大,为了进一步提高该反应的效率和实用性,我们设计开发了如图 5所示的aza-Wittig/Strecker一锅法不对称串联反应.不用分离提纯aza-Wittig反应生成的N-Boc靛红亚胺,直接进行下一步不对称Strecker反应时,在相同的反应条件下同样能以中等的产率和良好到优秀的对映选择性得到相应的产物14[35].虽然与单步Strecker反应相比,该串联反应产物的对映选择性稍有降低,但这却是一种利用现场生成的酮亚胺来发展串联不对称Strecker反应的全新策略,无需预先制备酮亚胺就可以实现手性α-季碳氨基酸衍生物的合成,从而可以大大地减少实验操作和分离纯化过程所带来的人力、时间和各种材料的消耗,并且有效地降低了产率损失.
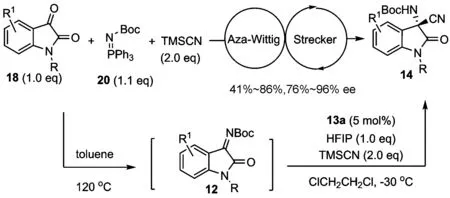
图 5 一锅法串联aza-Wittig/Strecker反应
利用这一方法,我们首次成功实现了具有止痛药效的化合物 Spirohydantoin I (8)的不对称催化全合成(图 6)[35].

图 6 Spirohydantoin I 的不对称全合成
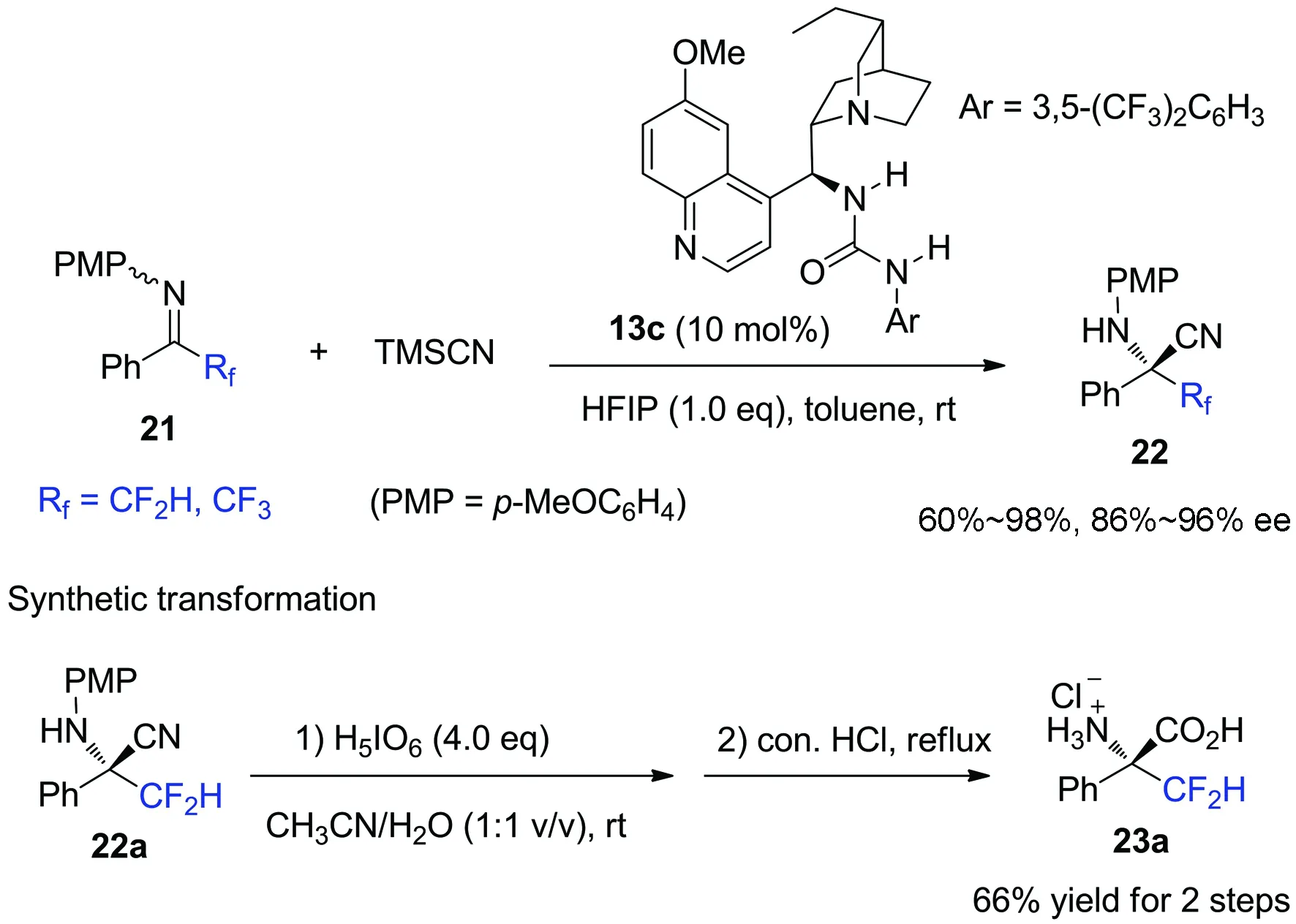
图 7 -氟代烷基酮亚胺的不对称Strecker反应
进一步研究发现,酮亚胺21中的氟原子对反应的对映选择性有强烈的影响(图8)[36].在相同条件下,非氟代酮亚胺只能以97%的收率得到消旋季碳胺基氰24;而α-CF3和CF2H取代的氟代酮亚胺则能分别以94%和87%的对映选择性得到相应的手性产物22a和22b.根据观察到的强烈氟效应,我们与四川大学王欣教授合作,从理论计算结果和实验数据,提出了氟代酮亚胺与(硫)脲催化剂的一种新型的识别模式(图8).对于甲基酮亚胺24而言,催化剂硫脲通过与亚胺的氮原子形成双氢键作用活化底物;而对于三氟甲基酮亚胺22a,计算表明硫脲的双氢键给体通过分别与底物的氮原子和α-碳上的氟原子形成双氢键作用更有利于活化底物.这不仅是对二氟甲基取代的酮亚胺的首例不对称催化亲核加成反应,也是C—F…H—N相互作用在不对称催化反应中影响反应活性和对映选择性的首例报道[37-40].
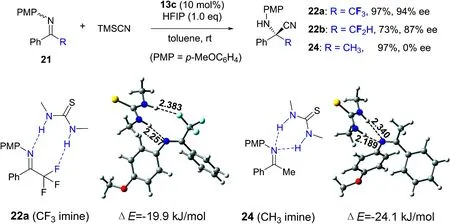
图 8 -氟代烷基酮亚胺不对称Strecker反应的氟效应
4 α-酮酸酯衍生的酮亚胺的亲核加成反应
α-酮酸酯衍生的酮亚胺与亲核试剂的不对称亲核加成反应是合成手性α-季碳氨基酸的另一种重要策略[41-47],该策略的优势在于可以使用多种不同结构的亲核试剂,从而合成结构多样性的α-季碳氨基酸衍生物.
2014年,我们发现利用1~5 mol%的DBU作为催化剂,可以高效催化硝基甲烷对α-酮酸酯衍生的酮亚胺的aza-Henry反应.在该条件下,芳基取代酮酸酯衍生的N-Ts亚胺25和靛红衍生的N-Boc酮亚胺12均可以取得良好到优秀的产率,得到相应的 α-胺基-β2,2-氨基酸衍生物.初步的条件考察发现,使用双功能叔胺氢键给体催化剂26a可以催化该反应取得高达71%的对映选择性 (图9)[48].

图 9 α-酮酸酯衍生酮亚胺的aza-Henry反应
随后,我们对硝基烷烃与N-Boc 靛红酮亚胺12的不对称aza-Henry 反应进行了深入研究,发现金鸡纳碱衍生的叔胺-酚羟基双功能催化剂26b可以很好地催化该反应,以66%~91%的对映选择性得到相应的3-胺基氧化吲哚28.而且,硝基乙烷和硝基丙烷在相同的条件下也能取得良好的对映选择性,但其非对映选择性还有待进一步提高(图 10)[49].
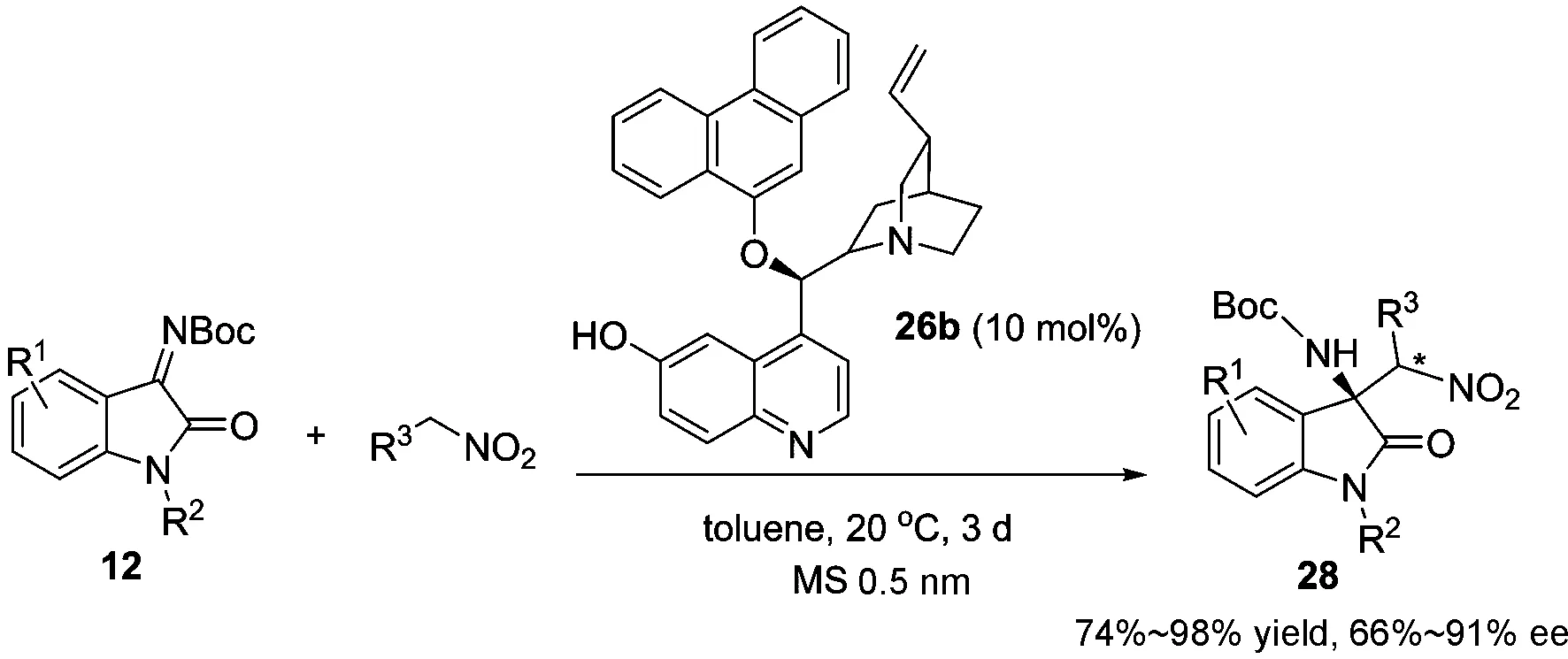
图 10 N-Boc靛红酮亚胺的不对称aza-Henry反应
与此同时,我们还发展了含有丙二酸酯结构的靛红酮亚胺29的不对称分子内的6π电环化反应.研究发现,在10 mol%奎宁衍生的双功能硫脲催化剂13d的作用下,该反应能以良好到优秀的产率和对映选择性得到含有氧化吲哚骨架的螺环吲哚啉类化合物30(图 11)[50].对照试验表明,催化剂13d中硫脲的协同活化作用非常关键,即使只是硫脲的一个N—H键被甲基保护后,反应也根本不能进行.基于此,我们推测了如图 11所示的双功能协同活化的反应过渡态.值得一提的是,这是首次利用不对称6π电环化反应来构建四取代碳手性中心的报道.
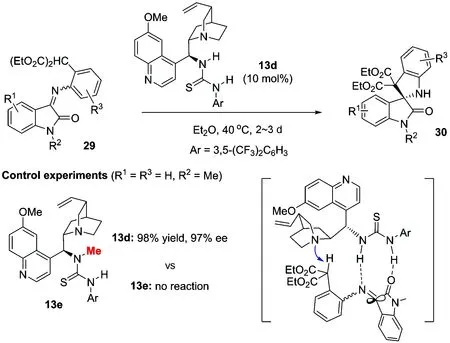
图 11 靛红酮亚胺29的不对称6π电环化反应
在此基础上,结合钯催化,布朗斯特酸催化和双功能叔胺-氢键给体催化,我们实现了首例不对称三重接力催化.利用不对称6π电环化反应作为关键步骤,从简单原料出发,经过硝基还原/亚胺形成/6π电环化三步串联反应,成功实现了30的快速合成 (图 12)[50].该串联反应底物普适性广,反应条件温和,为螺环化合物的高效构建提供了一种新方法;也为新型不对称催化串联反应的发展提供了一种新思路.

图 12 一锅法硝基还原/亚胺形成/6π电环化串联反应
最近,我们也报道了利用氢化奎宁衍生的脲催化剂13c作为催化剂,实现了氟代烯醇硅醚与环状N-磺酰基酮亚胺的不对称Mukaiyama-Mannich反应,以良好到优秀的产率和对映选择性得到具有二氟烷基取代季碳手性中心的α-氨基酸衍生物34(图 13)[51].该体系不仅适用于二氟烯醇硅醚,而且对单氟烯醇硅醚也同样兼容,用于合成相应的含有2个连续季碳手性中心的苯并噻唑类化合物.所得产物可以方便地进行转化,如通过Baeyer-Villiger氧化可以得到相应的二酯类化合物35;而经胺解环化可以制备具有螺环结构的苯并噻唑类化合物36.

图 13 氟代烯醇硅醚与酮亚胺的不对称Mannich反应
需要说明的是,在该反应研究过程中,我们也同样观察到了明显的氟取代效应(图 14).例如,将二氟烯醇硅醚33a换为简单非氟代烯醇硅醚37时,反应的活性和立体选择性均大幅度下降;而使用二氯烯醇硅醚40时,未观察到目标产物的生成.此外,直接使用相应的二氟甲基酮39时,反应同样不能发生,这也说明使用氟代烯醇硅醚来发展不对称催化反应,进而引入氟代烷基的重要性和必要性[52-57].
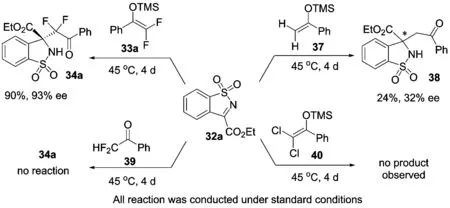
图 14 不对称Mannich反应中的氟效应
5 前手性氧化吲哚的不对称胺化反应
利用前手性羰基化合物与不同亲电胺化试剂的不对称胺化反应也是合成手性α-季碳氨基酸的一种有效策略[43].通过选择不同结构的前手性羰基化合物可以有效地构建结构多样性的手性α-季碳氨基酸.
鉴于我们小组对3,3-双取代氧化吲哚骨架的研究兴趣,开展了基于前手性3-取代氧化吲哚的一系列不对称亲电胺化反应来高效合成手性3-胺基季碳氧化吲哚这一特殊的α-季碳氨基酸衍生物.例如,早在2009年, 我们发现利用(QD)2PYR43作为催化剂,可以高效催化氮上未保护的3-烷基氧化吲哚41与偶氮二甲酸二异丙酯42的不对称胺化反应.该反应能以优秀的产率和对映选择性得到相应的胺基季碳氧化吲哚44(图15)[58].与此同时,文献[59]也实现了该胺化反应.
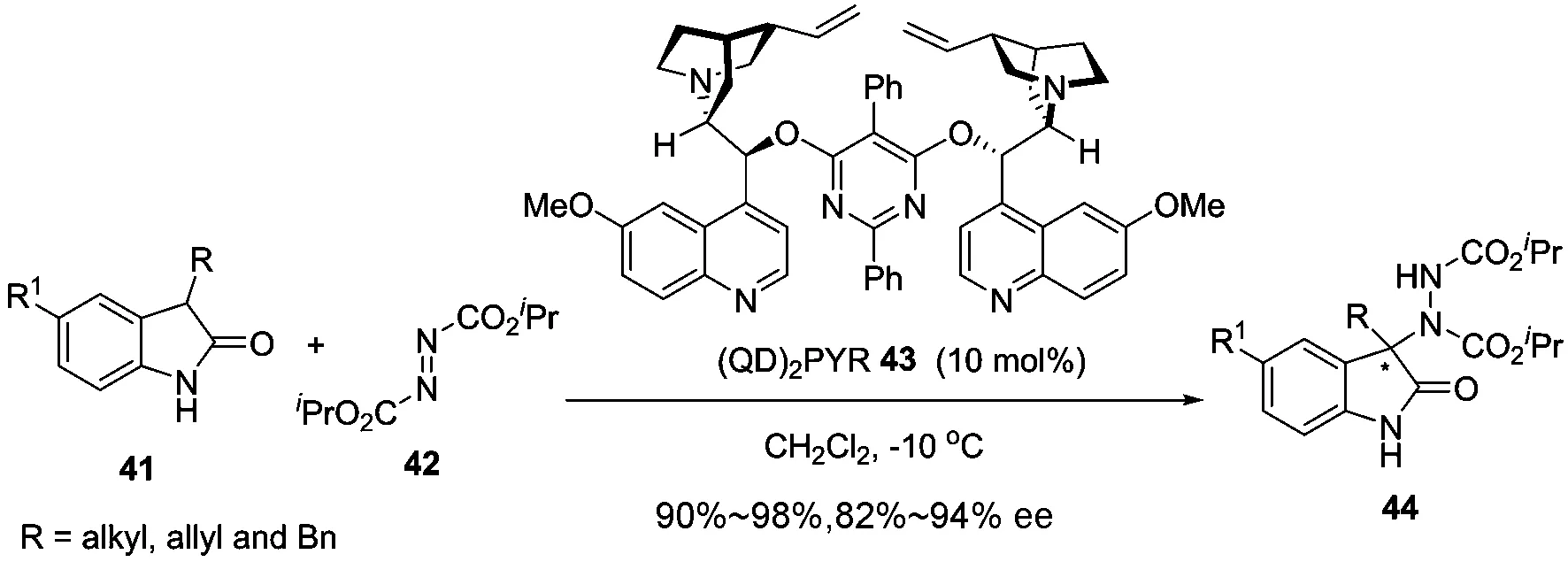
图 15 3-烷基氧化吲哚与偶氮二异丙酯的不对称胺化反应
尽管我们可以在温和条件下实现3-烷基取代氧化吲哚的不对称胺化反应,但由于该反应使用了偶氮二甲酸异丙酯42作为亲电胺化试剂,使得所得产物较难转化,因此,设想能否利用更易转化的偶氮二甲酸叔丁酯45(DBAD)作为胺化试剂,从而使所得产物可以方便地进行衍生化.为了成功实现该胺化反应,我们设想利用叔胺-氢键给体双功能催化剂来同时活化2个反应底物,使得反应类似于分子内的反应,更易进行.在这样一个设想下,我们发现,手性双功能硫脲催化剂13d可以高效催化氮上未保护的3-取代的氧化吲哚41与 DBAD45的不对称胺化反应,以良好到优秀的产率和对映选择性得到3-胺基氧化吲哚46.该反应不仅底物普适性非常广,无论3-芳基或3-烷基取代的氧化吲哚都可以取得优秀的对映选择性,而且所得产物可以方便地转化为具有重要应用价值的3-胺基氧化吲哚47(图16)[59].
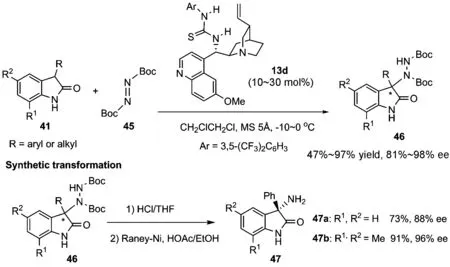
图 16 3-取代氧化吲哚与偶氮二甲酸叔丁酯的胺化反应
在实现前手性3-烷基或3-芳基取代氧化吲哚41与DBAD45的不对称亲电胺化反应后,我们还进一步报道了3位杂原子取代的氧化吲哚与偶氮二甲酸叔丁酯的不对称亲电胺化反应,从而成功合成了一系列3,3-双子原子取代的氧化吲哚类化合物.通过条件优化发现,利用 (DHQD)2PYR和β-ICD作为催化剂可以分别高效地催化3位含氧取代氧化吲哚48和3位含硫取代的氧化吲哚50与DBAD的不对称胺化反应,以中等到优秀的产率和对映选择性得到相应的化合物49和51(图17)[60].这些产物也非常有用,比如可以用于结构新颖的3位双杂原子取代的螺环氧化吲哚类化合物53的合成.

图 17 3-杂原子取代氧化吲哚与偶氮二甲酸叔丁酯的不对称胺化反应
6 α-取代硝基乙酸酯的不对称官能团化反应
α-氮杂羧酸衍生物如α-取代甘氨酸衍生物、噁唑啉酮类化合物、α-取代硝基乙酸酯类化合物等是合成α-季碳氨基酸的常用前体.由于该类化合物中α碳上含氮基团的拉电子诱导效应使得其α位氢酸性增强,因此较容易被攫氢活化,从而可作为一类活性较强的亲核试剂来设计不同的反应.其中,我们小组对利用α-取代硝基乙酸酯来合成手性α-季碳氨基酸这一策略比较感兴趣.
基于此,2012年,我们利用手性叔胺-酚羟基双功能催化剂β-ICD,成功实现了α-取代硝基乙酸酯54与偶氮二甲酸叔丁酯45的不对称亲电胺化反应(图18)[61],发现以1,1,2,2-四氯乙烷或二氯甲烷为反应溶剂,在-20或-40oC的低温条件下,α-脂肪族取代的硝基乙酸酯可以取得良好到优秀的产率和对映选择性.但在相同的反应条件下,α-芳香族取代的硝基乙酸酯的反应结果还有待进一步提高.所得目标产物经过简单的转化,即可以合成含有α-季碳氨基酸结构的四取代酰肼类化合物.

图 18 α-取代硝基乙酸酯与偶氮二甲酸叔丁酯的不对称胺化反应
此外,我们还报道了3-取代硝基乙酸酯的不对称羟甲基化反应.研究发现,在去甲基奎宁定56的催化作用下,利用多聚甲醛作为羟甲基化试剂,最高能取得71%的对映选择性 (图19)[62].

图 19 α-取代硝基乙酸酯与多聚甲醛的不对称羟甲基化反应
7 结论与展望
我们小组自成立以来一直关注α-季碳氨基酸的不对称催化合成研究.迄今为止,已利用酮亚胺的不对称Strecker反应、α-酮酸酯衍生的酮亚胺的不对称亲核加成反应、前手性氧化吲哚的不对称胺化反应以及α-硝基乙酸酯的不对称官能团化反应这4种策略,发展了一系列不对称催化新反应来结构多样性合成手性α-季碳氨基酸类衍生物.这些研究工作不仅为手性α-季碳氨基酸的高效合成提供了一些新方法,也在此过程中发展了一类新型的膦酰胺-叔胺双功能催化剂,发现了一些新型识别活化模式;这为以后化学家们研究类似工作提供了借鉴.
尽管如此,我们认为不对称催化合成α-季碳氨基酸仍然具有很大的发展空间.现有的方法往往催化剂的用量较高(需要10 mol%或以上),反应底物的多样性还不够,且已有方法的原子经济性较低.因此,继续深入研究并发展新型催化剂或催化模式来改善现有反应的效果,以及发展新型的不对称催化反应来合成结构更为丰富的手性α-季碳氨基酸,仍然是今后需要继续努力的方向.与其它学科合作,进一步揭示这些结构新颖的α-季碳氨基酸的实际应用,也是需要关注的一个方向.
[1] CATIVIELA C, DAZ-DE-VILLEGAS M D. Stereoselective synthesis of quaternary α-amino acids[J]. Tetrahedron:Asymmetry,1998,9:3517-3599.
[2] CATIVIELA C, DíAZ-DE-VILLEGAS M D. Stereoselective synthesis of quaternary α-amino acids. Part 2. Cyclic compounds[J]. Tetrahedron:Asymmetry,2000,11:645-732.
[3] TAKAHASHI A, KURASAWA S, IKEDA D, et al. Altemicidin, a new acaricidal and antitumor substance. I. Taxonomy, fermentation, isolation and physico-chemical and biological properties[J]. J Antibiotics,1989,42(11):1556-1561.
[4] NICOLAOU K C, SCHLAWE D, KIM D W, et al. Total synthesis of halipeptins:isolation of halipeptin D and synthesis of oxazoline halipeptin analogues[J]. Chemistry,2005,11(21):6197-6211.
[5] KODA Y, TAKAHASHI K, KIKUTA Y, et al. Similarities of the biological activities of coronatine and coronafacic acid to those of jasmonic acid[J]. Phytochemistry,1996,41(1):93-96.
[6] BLECHERT S, BOCKELMANN C, FÜLEIN M, et al. Structure-activity analyses reveal the existence of two separate groups of active octadecanoids in elicitation of the tendril-coiling response of Bryonia dioica, Jacq[J]. Planta,1999,207(3):470-479.
[7] MELOTTO M, UNDERWOOD W, HE S Y. Role of stomata in plant innate immunity and foliar bacterial diseases[J]. Annual Review of Phytopathology,2008,46(1):101-122.
[8] MIYAKE Y, KOZUTSUMI Y, NAKAMURA S, et al. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/Myriocin[J]. Biochemical & Biophysical Research Communications,1995,211(2):396-403.
[9] WANG B, LIN G Q. A flexible common approach to α-substituted serines and alanines:diastereoconvergent syntheses of sphingofungins E and F†[J]. European Journal of Organic Chemistry,2009,2009(29):5038-5046.
[10] OCHI M, KAWASAKI K, KATAOKA H, et al. AG-041R, a gastrin/CCK-B antagonist, stimulates chondrocyte proliferation and metabolism in vitro[J]. Biochemical & Biophysical Research Communications,2001,283(5):1118-1123.
[11] BEY P, GERHART F, VAN D V, et al. Alpha-(Fluoromethyl)dehydroornithine and alpha-(fluoromethyl)dehydroputrescine analogues as irreversible inhibitors of ornithine decarboxylase[J]. J Medicinal Chemistry,1983,26(11):1551-1556.
[12] SAARI W S, HALCZENKO W, COCHRAN D W, et al. 3-Hydroxy-alpha-methyltyrosine progenitors: synthesis and evaluation of some (2-oxo-1,3-dioxol-4-yl)methyl esters[J]. J Medicinal Chemistry,1984,27(6):713-717.
[13] HOROSZOK L, LEUNG C, TOMASZEWSKI M, et al. Utilisation de spiro[imidazolidine-4,3′-indole]2,2′,5′(1h)triones pour le traitement de troubles associes au recepteur 1 de vanilloide:WO, WO 2007091946 A1[P]. 2007.
[14] KAUL R, BALARAM P. Stereochemical control of peptide folding[J]. Bioorganic & Medicinal Chemistry,1999,30(27):105-117.
[15] TANAKA M. Design and synthesis of chiral alpha,alpha-disubstituted amino acids and conformational study of their oligopeptides[J]. Chemical & Pharmaceutical Bulletin,2007,38(30):349-358.
[16] TONIOLO C, CRISMA M, FORMAGGIO F, et al. Control of peptide conformation by the Thorpe-Ingold effect (C alpha-tetrasubstitution)[J]. Biopolymers,2001,60(6):396-419.
[17] SCHILLER P W, WELTROWSKA G, NGUYEN T M, et al. Conformational restriction of the phenylalanine residue in a cyclic opioid peptide analogue:effects on receptor selectivity and stereospecificity[J]. J Medicinal Chemistry,1991,34(10):3125-3132.
[18] BURGESS A W. Designing amino acids to determine the local conformations of peptides[J]. Proc Natl Acad Sci USA,1994,91:2649-2653.
[19] PAUL P K C, SUKUMAR M, BARDI R, et al. Stereochemically constrained peptides. Theoretical and experimental studies on the conformations of peptides containing 1-Aminocyclohexanecarboxylic acid[J]. Cheminform,1987,18(6):6363-6370.
[20] KARLE I L, KAUL R, RAO R B, et al. Stereochemical analysis of higher α,α-dialkylglycine containing peptides. Characterization of local helical conformations at Dipnpylglycine Residues and observation of a novel hydrated multple β-turn structure in crystals of a golycine rich peptide[J]. J Am Chem Soc,1997,119:12048-12054.
[21] WANG J, LIU X, FENG X. Asymmetric Strecker reactions[J]. Chemical Reviews,2011,111(11):6947-6983.
[22] 唐然肖,李云鹏,李越敏,等. 有机小分子催化的不对称Strecker反应研究进展[J]. 有机化学,2009,29(7):1048-1058.
[23] VACHAL P, JACOBSEN E N. Enantioselective catalytic addition of HCN to ketoimines. Catalytic synthesis of quaternary amino acids[J]. Organic Letters,2000,31(27):867-870.
[24] VACHAL P, JACOBSEN E N. Structure-based analysis and optimization of a highly enantioselective catalyst for the Strecker reaction[J]. Cheminform,2002,124(34):10012-10014.
[25] MASUMOTO S, USUDA H, SUZUKI M, et al. Catalytic enantioselective Strecker reaction of ketoimines[J]. Cheminform,2003,34(36):5634-5635.
[26] WANG J, HU X, JIANG J, et al. Asymmetric activation of tropos 2,2′-Biphenol with cinchonine generates an effective catalyst for the asymmetric Strecker reaction ofN-Tosyl-Protected aldimines and ketoimines[J]. Angewandte Chemie,2007,46(44):8468-8470.
[27] SHEN K, LIU X, CAI Y, et al. Facile and efficient enantioselective Strecker reaction of ketimines by chiral sodium phosphate[J]. Chemistry,2009,15(24):6008-6014.
[28] DR J W, WANG W, LI W, et al. Asymmetric cyanation of aldehydes, ketones, aldimines, and ketimines catalyzed by a versatile catalyst generated from cinchona alkaloid, achiral substituted 2,2′-Biphenol and tetraisopropyl titanate[J]. Chemistry - A European Journal,2009,15(43):11642-11659.
[29] ABELL J P, YAMAMOTO H. Dual-Activation asymmetric Strecker reaction of aldimines and ketimines catalyzed by a tethered bis(8-quinolinolato) Aluminum complex[J]. Cheminform,2010,41(10):15118-15119.
[30] RUEPING M, SUGIONO E, MORETH S. Metal-free, enantioselective Strecker reactions catalyzed by chiral BINOL and TADDOL catalysts[J]. Advanced Synthesis & Catalysis,2010,349(4/5):759-764.
[31] ENDERS D, GOTTFRIED K, RAABE G. Organocatalytic enantioselective Strecker synthesis of α-Quaternary α-Trifluoromethyl amino acids[J]. Advanced Synthesis & Catalysis,2011,42(18):3147-3152.
[32] ZHOU F, LIU Y L, ZHOU J. Catalytic asymmetric synthesis of oxindoles bearing a tetrasubstituted stereocenter at the C-3 position[J]. Advanced Synthesis & Catalysis,2010,352(9):1381-1407.
[33] YU J S, ZHOU F, LIU Y L, et al. A journey in the catalytic synthesis of 3-substituted 3-aminooxindoles[J]. Synlett,2015,26(18):2491-2504.
[34] LIU Y L, ZHOU F, CAO J J, et al. A facile method for the synthesis of oxindole based quaternary alpha-aminonitriles via the Strecker reaction.[J]. Organic & Biomolecular Chemistry,2010,8(17):3847-3850.
[35] LIU Y L, ZHOU J. Organocatalytic asymmetric Cyanation of Isatin derived N-Boc Ketoimines[J]. Cheminform,2013,44(37):4421-4423.
[36] LIU Y L, SHI T D, ZHOU F, et al. Organocatalytic asymmetric Strecker reaction of Di- and trifluoromethyl ketoimines. Remarkable fluorine effect[J]. Organic Letters,2011,13(15):3826-3829
[37] CAHARD D, BIZET V. The influence of fluorine in asymmetric catalysis[J]. Chemical Society Reviews,2014,43(1):135-147.
[38] ZHAO X, WANG X Z, JIANG X K, et al. Hydrazide-based quadruply hydrogen-bonded heterodimers. Structure, assembling selectivity, and supramolecular substitution[J]. J the American Chemical Society,2003,125(49):15128-15139.
[39] LIU Y H, ZHANG L, XU X N, et al. Intramolecular C—H…F hydrogen bonding-induced 1,2,3-triazole-based foldamers[J]. Org Chem Front,2014,1:494-500.
[40] YU J S, LIU Y L, TANG J, et al. Highly efficient “On Water” catalyst-free nucleophilic addition reactions using difluoroenoxysilanes:dramatic fluorine effects [J]. Angew Chem Int Ed,2014,53(36):9512-9516.
[41] METZ A E, KOZLOWSKI M C. Recent advances in asymmetric catalytic methods for the formation of acyclic α,α-disubstituted α-amino acids[J]. J Organic Chemistry,2015,80(1):1-7.
[42] LIU Y L, ZHOU L. Catalytic asymmetric Strecker reaction:bifunctional chiral tertiary amine/Hydrogen-Bond donor catalysis joins the field[J]. Synthesis,2015,47(9):1210-1226;
[43] ZHOU F, LIAO F M, YU J S, et al. Catalytic asymmetric electrophilic amination reactions To form Nitrogen-Bearing tetrasubstituted Carbon stereocenters[J]. Synthesis,2014,46(22):2983-3003.
[44] ZHOU J. Catalytic asymmetric synthesis of Cα-tetrasubstituted α-amino acids[J]. Organic Chem Curr Res,2014,46(34):3,e136,doi:10.4172/2161-0401.1000e136;
[45] WANG J, LIU X, FENG X. Asymmetric Strecker reactions[J]. Chemical Reviews,2011,111(11):6947-6983.
[46] MERINO P, TEJERO T, HERRERA R P. Organocatalyzed Strecker reactions[J]. Tetrahedron,2009,65(7):1219-1234.
[47] VOGT H, BR SE S. Recent approaches towards the asymmetric synthesis of alpha,alpha-disubstituted alpha-amino acids[J]. Organic & Biomolecular Chemistry,2007,5(3):406-430.
[48] WANG Y H, LIU Y L, CAO Z Y, et al. An organocatalytic addition of Nitromethane to activated ketimines[J]. Asian J Org Chem,2014,3(4):429-432.
[49] 王雨卉,曹中艳,牛艳霏,等. 高对映选择性的有机催化的硝基烷烃对N-Boc靛红亚胺的不对称aza-Henry反应[J]. 化学学报,2014,72(7):867-872.
[50] YIN X P, ZENG X P, LIU Y L, et al. Asymmetric triple relay catalysis:enantioselective synthesis of spirocyclic indolines through a One-Pot process featuring an asymmetric 6π electrocyclization[J]. Angewandte Chemie,2014,53(50):13740-13745.
[51] YU J S, ZHOU J. Organocatalytic enantioselective Mukaiyama—Mannich reaction of fluorinated enol silyl ethers and cyclicN-sulfonyl ketimines[J]. Org Chem Front,2016,47(28):298-303.
[52] LIU Y L, ZHOU L. Organocatalytic asymmetric cyanation of isatin derived N-Boc ketoimines[J]. Chem Commun,2013,49(39):4421-4423;
[53] LIU Y L, LIAO F M, NIU Y F, et al. Highly stereoselective construction of adjacent tetrasubstituted carbon stereogenic centres via organocatalytic Mukaiyama-aldol reaction of monofluorinated silyl enol ethers to isatins[J]. Org Chem Front,2014,1(7):742-747.
[54] YU J S, ZHOU J. A highly efficient Mukaiyama-Mannich reaction of N-Boc isatin ketimines and other active cyclic ketimines using difluoroenol silyl ethers catalyzed by Ph3PAuOTf[J]. Cheminform,2015,13(45):10968-10972.
[55] YU J, LIAO F, GAO W, et al. Michael addition catalyzed by chiral secondary amine phosphoramide using fluorinated silyl enol ethers:formation of quaternary Carbon stereocenters[J]. Angewandte Chemie,2015,54(25):7381-7385.
[56] LIAO F M, CAO Z Y, YU J S, et al. Highly stereoselective gold-catalyzed coupling of diazo reagents and fluorinated enol silyl ethers to tetrasubstituted alkenes [J]. Angew Chem Int Ed,2017,129(9):2459-2463.
[57] LIAO F M, GAO X T, HU X S, et al. A general and efficient Lewis acid catalysed Mukaiyama-aldol reaction of difluoroenoxysilanes and ketones[J]. Sci Bull,2017,62(22):1504-1509.
[58] QIAN Z Q, ZHOU F, DU T P, et al. Asymmetric construction of quaternary stereocenters by direct organocatalytic amination of 3-substituted oxindoles[J]. Cheminform,2010,41(13):6753-6755.
[59] ZHOU F, DING M, LIU Y L, et al. Organocatalytic asymmetric α-amination of unprotected 3-aryl and 3-aliphatic substituted oxindoles using Di-tert-butyl azodicarboxylate[J]. Adv Syn Catal,2011,353(16):2945-2952.
[60] ZHOU F, ZENG X P, WANG C, et al. Organocatalytic asymmetric synthesis of 3,3-disubstituted oxindoles featuring two heteroatoms at C3 position[J]. Chem Commun,2013,49(20):2022-2024.
[61] JI C B, LIU Y L, ZHAO X L, et al. Direct amination of α-substituted nitroacetates using di-tert-butyl azodicarboxylate catalyzed by Hatakeyama’s catalyst β-ICD[J]. Org Biomol Chem,2012,10(6):1158-1161.
[62] JI C B, LIU Y L, CAO Z Y, et al. Hydroxymethylation of α-substituted nitroacetates[J]. Tetrahedron Lett,2011,52(46):6118-6121.
