肿瘤血管抑制剂Combretastatins的重要中间体CA4酸质量研究
2018-05-14孙嘉茵李宇茜熊小平樊淑宏罗婷婷雍志全徐小平
孙嘉茵 李宇茜 熊小平 樊淑宏 罗婷婷 雍志全 徐小平

摘要:对Combretastatin系列化合物的重要中间体CA4酸(CA4s)理化特征、有关物质、残留溶剂和含量测定的方法进行系统研究。通过IR、UV、MS、NMR等确定本品的化学结构。采用HPLC法检测有关物质,条件为:色谱柱C18(150mm×4.60mm,5μm),流动相(体积比):0.1%甲酸(氨水调解pH至6.8)-甲醇-乙腈(75:22:3),检测波长为220nm;采用HS-GC法检测残留溶剂,条件为:PEG20M(30m×0.53mm,1μm)的石英毛细管柱,柱温80℃,检测器温度250℃(FID),进样口温度200℃,流量4.0mL/min,分流比:10:1;采用LC-MS鉴定本品有关物质;以剩余滴定法测定本品的含量。结果表明:CA4酸为难溶于水的浅黄色结晶性粉末,其化学结构与2-(3,4,5-三甲氧苯基)-3-[3-羟基-4-甲氧苯基卜丙烯酸相吻合,有6个杂质组分,经鉴定杂质5和杂质2为三甲氧基苯乙酸和异香草醛;杂质6为z式CA4酸,受光照影响发生较大的变化反映出顺反异构的特征;残留溶剂乙醇量小于0.08%。CA4酸为Combretastatinl类化合物的关键中间体,难溶于水,分子中的羧基可与碱性化合物作用生成可溶性的盐,可突破该类化合物难溶性瓶颈,提高该类化合物抑制肿瘤血管的效率,是一个具有较高价值Combretastatins中间体或先导化合物。
关键词:CA4酸;抗肿瘤药;高效液相色谱;气相色谱
文献标志码:A
文章编号:1674-5124(2018)02-0041-07
0引言
Combretastatin类化合物是近年来发现的一类肿瘤血管抑制剂,其二苯乙烯母核的结构发现于南非灌木柳树Combretumcaffrum的树皮中,是一类具有顺式二苯乙烯结构的天然产物,其抗肿瘤作用机理与秋水仙碱结构类似,是通过竞争抑制与微管蛋白相结合,从而发挥其抗有丝分裂的作用,达到抑制微管形成,阻滞细胞增殖以及抗血管活性的效果。在这类化合物中,Combretastatin A-4(CA4)具有最佳的微管抑制效果及体外细胞毒性,且对有异常结构特征和功能的肿瘤血管具有靶向抑制性,对正常血管作用较小,因而全身毒性较小且不易产生耐药性,因而备受关注。但由于CA4具有水溶性较差且难以保持其有效构型等缺点,限制了其临床应用。对该类化合物结构修饰并改善其水溶性是解决它们最终抗癌作用的关键,包括骨架及基團修饰和合成氨基糖类衍生物等。近年,国内外相继开发出的CA4磷酸盐前药CA4P已进入II、III期临床试验。
本文在CA4和CA4P研究的基础上,发掘一种具有酸性结构的中间体——CA4酸(CA4s),可以通过成盐产生具有良好水溶性的化合物,从而突破这类化合物的瓶颈。采用3,4,5-三甲氧基苯乙酸和异香草醛在乙酸酐和三乙胺中进行Perkin缩合反应,经浓盐酸处理,并经无水乙醇重结晶精制,得到(E)-3-(3-羟基-4-甲氧基苯基)-2-(3,4,5-三甲氧基苯基)-丙烯酸(即CA4s,路线见图1)。近期,CA4s与有机胺结合产生的DX1002作为肿瘤血管抑制剂已经申请国家一类创新药临床试验,而CA4s的质量控制成为DX1002质量的关键,建立中间体CA4s的质量标准具有非常重要的意义,由于CA4s为首次开发,暂无直接的研究报道,本文根据CA4s合成特点,针对其化学结构、性质和质量特征进行系统研究,确证其化学结构、性状、种类,检查其有关物质、建立含量测定方法,为进一步深入研究开发DX1002系列化合物提供中间体的分析方法和物质基础。
1实验部分
1.1仪器和试剂
LC-10AT高效液相色谱仪(日本岛津),SPD-20A紫外/可见检测器(日本岛津),SePu3000色谱工作站(杭州普惠科技有限公司);Nicolet Impact 410型红外光谱仪(美国Nicolet仪器公司);UV-2300紫外-可见分光光谱仪(上海天美科学仪器有限公司);GC7890F气相色谱仪(FID,上海天美科学仪器有限公司);Varian UNITY INOVA型400核磁共振谱仪(美国Nicolet仪器公司);轨道阱高分辨质谱系统(LTQ Orbitrapelite美国热电分析仪器公司);pHS-3C型酸度计(成都方舟科技开发公司);Sartorious BT 125D十万分之一电子(德国Sartorius);SHANGPING FA2004万分之一电子天平(上海天平仪器厂);KL10260D型超声波清洗器(上海荆和分析仪器有限公司);DHG-9140A电热恒温鼓风干燥器(上海精宏实验设备有限公司);SHZ-D(III)循环水式真空泵(成都市予华伟业仪器有限公司);超纯水机(成都品成科技有限公司);旋涡振荡器(江苏海门麒麟医用仪器厂)。
CA4s样品及其顺式结构样品(自制,东莞达信生物技术有限公司);异香草醛对照品(Adamas Reagentco.,Ltd.,批号:P1097634)和3,4,5-三甲氧基苯乙酸对照品(Adamas Reagentco.,Ltd.,批号:P03109);氢氧化钠(AR,天津市科密欧化学试剂有限公司);浓硫酸(AR,成都市天华科技股份有限公司);甲醇(HPLC,天津科密欧化学试剂有限公司);乙腈(HPLC,进口分装);氨水(AR,广东光华科技股份有限公司);无水乙醇(HPLC,天津科密欧化学试剂有限公司);水为纯水机自制超纯水。
1.2方法
1.2.1CA4s的结构确证
分别通过核磁共振(NMR)、质谱(MS)、红外分光光度法(IR)和紫外分光光度法考察(CA4s)的结构。
1.2.2CA4s的理化性质测定
1)性状的考察。取洁净滤纸一张,挑取样品少许置于其上,平铺均匀,于自然光下观察。取3份样品适量,分别置于高温(60℃)、高湿(相对温度为92.5%)和光照(4500k)条件下1h,观察外观变化。
2)溶解度的考察。照ChP.2015凡例要求,称取样品适量,于(25±2)℃条件下,加入待考察的溶剂适量,每隔5min强力振摇30s,观察溶解情况。考察溶剂包括:水、甲醇、乙醇、丙酮、氯仿、DMSO、DMF。
3)pH的考察。取样品适量,加超纯水配制饱和溶液,测定pH。
4)酸、碱条件下紫外图谱。取样品适量,精密称定,分别加0.1mol/LhCl溶液和0.1mol/LnaOH溶液制成含量为20μg/mL的溶液,在波长200~400nm进行紫外扫描。
1.2.3有关物质的研究
1)色谱条件的确定
以CA4P分离色谱条件为基础,确定对CA4s的色谱条件为C18(150mm×4.60mm,5μm);流动相比例(体积比)为0.1%甲酸(pH 6.8)-甲醇-乙腈(75:22:3),柱温30℃,流量1mL/min。
2)方法学验证
照上述建立的CA4s有关物质HPLC检测条件进行了相关方法学验证,内容包括:1)专属性试验;2)检测限与定量限的测定;3)精密度试验(进样精密度、重复性精密度及中间精密度)。
3)重要有关物质的鉴定研究
根据CA4s合成工艺及其有关物质分离结果表明,本品除了来自原料的异香草醛、三甲氧基苯乙酸外,受温度和光照影响最明显的为本品主峰后的杂质峰,为此,采用LC/MS对杂质进一步鉴定。色谱条件为ODS(4.6mm×250mm)色谱柱:流动相0.1%甲酸(氨水调pH 6.8)-甲醇-乙腈=75:22:3,流量1mL/min,进样量为10μL,柱温40℃:质谱条件为安捷伦三重串联四级杆质谱,干燥气为氮气,气体温度:350℃,流量:11L/min,电喷雾离子源(ESI),毛细管电压:4kV,雾化气压力:40psi(1psi=6.895kPa),裂解电压:75V,离子化方式为正模式。碰撞能量:15V,细胞加速电压:3V,检测方式为多重反应监测(MRM)方法。
1.2.4残留溶剂的检查
根据CA4s的合成和精制工艺,确定本品残留溶剂为乙醇(bp.78.4℃)。参照ChP.2015通则0521气相色谱法和通则0861残留溶剂测定法,建立气相色谱法检测CA4s中的残留乙醇并进行方法学验证。
1)色谱条件的选择
本文对色谱柱SE-54(30m×0.32mm,0.25μm)、OV-624(30m×0.53mm,0.5μm)、OV-17(15m×0.53mm,1.0μm)、PEG(30m×0.53mm,1μm);分流比(200:30、200:25、200:20、200:20、200:15、200:10、200:5);流量3.5,4.0,4.5mL/min和柱温60,80,100,120℃、进样方式(直接进样、顶空进样)以及溶剂(DMF、DMSO)等条件做了筛选,以乙醇的理论塔板数、分离度、拖尾因子为指标进行考察。
2)线性与灵敏度
CA4s中残留乙醇约0.1%,因此设置线性范围为0.0067-1.0796mg/mL,进样浓度为0.0067,0.0135,0.0270,0.0540,0.1080,0.2699,0.5398,1.0796mg/mL。顶空后,依次精密量取上述溶液1mL分别注入气相色谱仪,记录色谱图。按信噪比法,在S/N≥10,确定定量限,在S/N≥3,确定检出限。
3)回收率
供试品溶液的配制:分别精密取本品9份(每份约1g)置于10mL容量瓶中,分为高、中、低3组。往低浓度组加入乙醇浓度为0.75mg/mL的对照品溶液0.8mL;往中浓度组加入乙醇浓度为0.75mg/mL对照品溶液1mL;往高浓度组加入乙醇浓度为0.75mg/mt的对照品溶液1.2mL,加入DMSO稀释至刻度,分别作为CA4s回收率80%、100%、120%的供试品溶液。
1.2.5含量测定研究
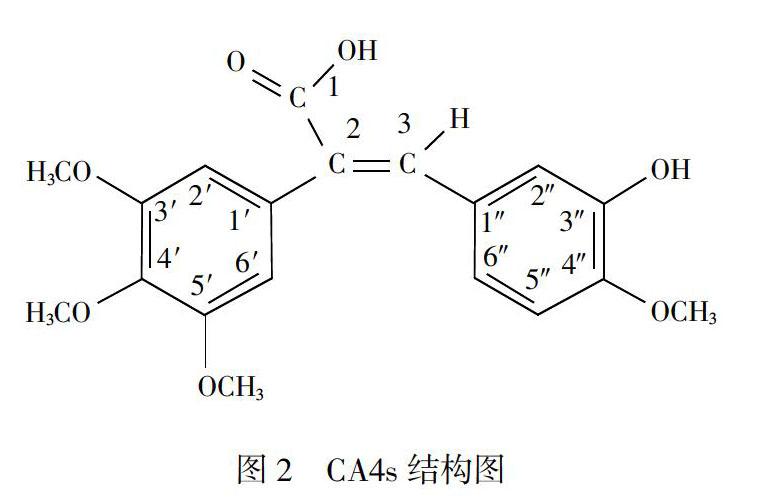
根据CA4s结构(见图2)中的羧基呈酸性的特点,试验将采用酸碱滴定法就其含量进行测定。由于CA4s水溶性极差,所以,在设计本品含量测定方法时,采用了以定量过量NaOH溶液溶解,然后用硫酸标准溶液滴定的剩余滴定法来实现。
1)滴定曲线的测定及指示剂的选择
取样品约100mg,精密称定,加入0.1mol/LnaOH滴定液25mL,摇匀溶解后加入50mL新沸水,用0.05mol/Lh2SO4滴定液滴定至溶液中有沉淀析出,并在滴定过程中,记录滴定过程的pH值变化,绘制滴定曲线。
2)精密度试验
重复性精密度:取同一批CA4s 6份,经含量测定,以CA4s的含量计算重复性精密度:中间精密度试验:取本品,分别由两个不同的实验员连续6d、每人测定6份样品含量,计算含量平均值和RSD。
2結果与讨论
2.1结构确认
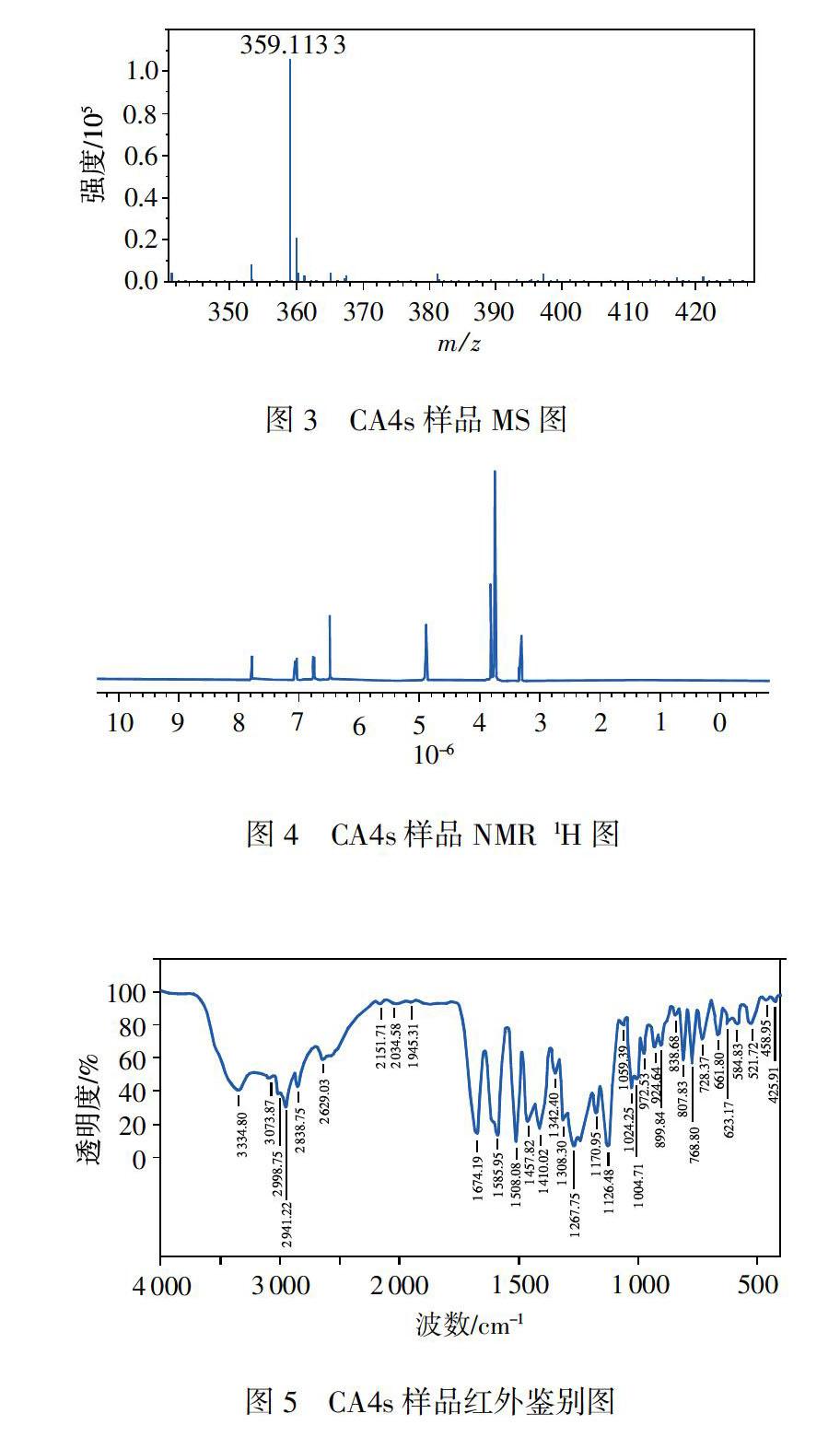
如图3所示,MS的分子离子峰(+m/z)为359,碎片峰与CA4s的分子量360的M-1吻合:NMR tH谱(图4)的氢谱的归属分别为:63.296(d,3H,4"--OCH3),δ3.753(d,9H,3,4,5-OCH3),δ6.484(s,2H,2,6'-H),δ6.545(s,1H,2"-H),δ6.676(d,1H,5"-H),δ6.788(d,1H,6"-H),δ7.707(s,1H,3-H),除两个活泼氢被D2O氘代,其他皆与结构吻合(具体编号见图2);IR光谱(图5)中波数3300,2900,1670,1600,1400cm-1等分别表现出结构中的羟基、甲基、羰基、甲氧基等,与CA4s分子结构中的相关基团吻合;UV光谱(图6)特征表明,本品具有共轭体系,受羧基和酚羟基的影响,B带和E带都发生长移现象,最大吸收为311nm,与CA4s的机构特征吻合。4大谱结果表明本品的结构为2-(3,4,5-三甲氧基-苯基)-3-(3-甲氧基2-羟基-苯基)丙烯酸。对比本品顺反式UV谱(图7)可知,由于羟基苯基在空间上影响到羧基的作用,使得311nm的最大吸收萎缩,并250nm的肩峰凸显,由此证明本品系羧基与羟苯基于异侧的反式或E式结构。即(E)2-(3,4,5'-三甲氧基-苯基)-3-(3”-甲氧基2"-羟基-苯基)丙烯酸。
2.2理化性质
试验结果表明,本品为浅黄色结晶性粉末;在高温和高湿条件下较稳定,光照时出现颜色加深。难溶于水,微溶于甲醇和丙酮,易溶于乙醇,是其难以以原型发挥活性的根源,饱和水溶液的pH为6.48,在酸性条件较稳定,在碱性条件下不稳定。
2.3有关物质研究
2.3.1色谱分离的影响因素
如图8所示,水相的pH对分离呈负相关,pH升高保留时间缩短:水相比例提高保留时间增加;柱温对本品色谱行为无明显影响。
2.3.2有关物质的鉴定
结合CA4s的合成途径,起始原料3,4,5-三甲氧基苯乙酸和异香草醛的色谱定位,以及LC/MS对本品杂质谱的分离和在线鉴定,分离出6个杂质组分,其中本品主峰前的杂质2为异香草醛、杂质5为3,4,5-三甲氧基苯乙酸。而杂质6与主峰分子质量相同,但保留时间相差甚远,结合紫外吸收图谱和核磁共振氢谱分析,确定杂质6为CA4s的顺式(Z式)异构体,结构如图9所示。
2.3.3检出限和定量限
按信噪比法,在S/N≥10和S/N≥3,确定CA4s的定量限为1ng、检出限为0.2ng。
2.3.4专属性试验
结果表明,酸破坏(4mol/LhCl破坏4h)和碱破坏(4mol/LnaOH破坏4h)条件下,供试品溶液杂质峰数量无明显增加,主峰也无明显降解;在氧化破坏(30%H2O2破坏4h)和高温破坏(160℃放置1h)条件下,杂质峰含量略有增加,主峰无明显降解。在光照破坏(4500Lx破坏4h)条件下,杂质峰数量和含量都有增加,杂质6含量增加尤为明显,主峰也显著降解。
2.3.5精密度
连续进样6针,各杂质峰面积RSD在2.27%~5.03%,表明此方法进样精密度良好。平行配制6份供试品溶液进样,以不加校正因子的自身稀释对照法计算各杂质含量,RSD在1.63%-4.67%之间,表明此方法重复性良好。连续3d,每天由两名实验员平行配制6份供试品溶液进样,以不加校正因子的自身稀释对照法计算各杂质含量,RSD在2.84%~5.07%,表明此方法中间精密度良好。
2.3.6有关物质的含量检查
用建立的方法对自制3批样品进行有关物质检查,3批样品中单个杂质含量均小于0.05%,总杂质含量小于0.5%,符合规定。
2.3.7有关物质研究小结
由于CA4s在水中几乎不溶,采用甲醇作为溶剂时,各杂质峰有关物质峰理论塔板数均较低,对称性差,且溶剂峰对保留时间短的杂质出峰影响很大。故采用甲醇溶解样品制成高浓度的储备液(约为5mg/mL),再用流动相作为溶剂,稀释制成0.5mg/mL的供试品溶液,采用这种方法制备的供试品溶液色谱图中,各杂质峰柱效有了显著提高。通过专属性试验可以看出,CA4s对酸性、碱性、氧化、高温条件耐受性较高,在光照的条件下容易发生构型反转,得到顺式结构的杂质6。因此在合成和储存过程中,应注意避光。
2.4残留溶剂
2.4.1色谱条件影响
与其他3种类型色谱柱相比较时,PEG毛细管色谱柱在柱效、分离度和峰对际性方面均有明显优势,并且分析时间合适;分流比对本品分析影响较大,随着分流比增大,出峰时间延迟,乙醇峰理论塔板数升高,与基质中的杂质峰分离度有所提高,但灵敏度有所下降,当分流比10:1时,既能保证乙醇峰有较高的柱效、分离度和对称性,保留时间也比较合适,故选择分流比10:1;不同流量对本品分析影响不大,流量4.0mL/min时,有较好的柱效、分离度、对称性,能保证残留溶剂乙醇的有效检出,故选择流量4.0mL/min;柱温对柱效和拖尾因子影响不大,但随着柱温的升高,乙醇峰的分离度显著下降,柱温80℃时,能保证残留溶剂乙醇的有效检出,且与空白溶剂中相关峰达到有效分离,故照国家药品标准,选择柱温80℃:顶空进样与直接进样相比,柱效和峰对称性等方面均有明显优势,顶空进样可避免基质影响和样品对色谱柱的污染,故选择顶空进样方式;溶剂DMF在乙醇峰前面有溶剂峰,影响乙醇的检出,故选择DMSO作为残留溶剂检查的样品溶剂。
2.4.2系统适应性条件
色谱柱为PEG毛细管色谱柱(30m×0.53mm,1μm),柱温80℃,检测器为氢火焰离子化检测器(FID),检测器温度250℃:进样口温度200℃;载气为氮气,流量为4.0mL/min,分流比10:1;顶空进样,顶空瓶温度80℃,平衡时间40min进样:进样体积1.0mL。
2.4.3线性与灵敏度
由乙醇峰高(H)对乙醇溶液浓度回归作图,获得乙醇的标准曲线方程为y=674724.8172x-2034.2,(r2=0.9993)。乙醇浓度在0.0067-1.0796mg/mL范围内,其线性关系良好。按信噪比法,在S/N≥10,确定定量限1.8μg/mL;在S/N≥3,确定检出限为0.6μg/mL。
2.4.4回收率与精密度
经过高、中、低3种浓度的回收率测定结果分别为98.06%(RSD为4.57%)、103.6%(RSD为1.73%)、93.63%(RSD为7.21%):进样精密度RSD为1.90%;重复性精密度测定残留溶剂含量为0.0739%(RSD为4.10%)。方法的准确度和精密度均良好。
2.4.53批样品的残留溶剂结果
用建立的方法对自制3批样品进行残留溶剂检查,3批样品中残留溶剂含量为0.0748%、0.0727%、0.0795%,符合规定。
2.5含量测定
2.5.1滴定曲线的考察
滴定曲线确定突跃范围为7.5~9.0,由于本品为难溶于水的酸性化合物,故实验设计本品预先溶于定量的NaOH溶液中,由HCl标准溶液回滴定剩余的碱液,經空白校正后,计算出本品的含量。根据溶液体系由碱到酸的滴定过程,试验选择了碱性指示剂酚酞指示终点,溶液由红色变为无色。
2.5.2CA4s的含量测定方法
取样品约300mg,精密称定,加入0.1mol/LnaOH滴定液25mL,充分摇匀溶解后加入50mL新沸水,加入酚酞指示剂2滴,用0.05mol/Lh2SO4滴定液滴定至溶液红色消失,且0.5min内颜色无改变,并将结果用空白试验校正。每1mLh2SO4(0.05mol/L)滴定液相当于36.04mgc19H20O7。
2.5.3精密度试验
重复性精密度测定结果为100.7%(RSD 0.08%):中间精密度测定结果为100.5%(RSD 0.11%),说明此方法精密度良好。
2.5.4 3批样品含量测定
用建立的方法对自制3批样品含量进行测定,3批样品含量分别为100.4%(RSD 0.057%)、100.6%(RSD 0.150%)、100.6%(RSD 0.057%)。
2.5.5容量法研究小结
采用酚酞作为指示剂时,溶液浓度对重点判断影响很大,浓度越大变色现象越不明显,经过多次对比试验,在碱溶液中加入50mL新沸水对溶液进行稀释后,能显著观察到变色现象,有利于减少终点误差。
3结束语
CA4s作为Combretastatin类化合物重要中间体,其质量将对终产物造成直接影响。本品的化学结构鉴定结果表明与CA4s的结构式吻合:所建立的HPLC杂质检查法、GC残留溶剂检查法和容量分析法能够有效检测出本品的有关物质、残留溶剂和含量,可为进一步研究CA4s提供良好的分析手段。
