波函数及其在稀土离子晶场参数从头计算中的应用
2018-05-11周清卿占生宝
闻 军,周清卿,占生宝
1 “量子力学”中波函数概念
量子力学中的波函数是对于一个体系(例如微观粒子或粒子体系)量子态的数学描述。它可表示为坐标空间中所有粒子位置坐标的函数,也可表示为动量空间中所有粒子动量的函数,两者之间互为傅里叶变换。波函数是一种具有复数值的几率幅。基于它,人们可以获得针对体系所做测量可能结果的几率。根据叠加原理,各波函数可以乘以相应复数,再相加构成新的波函数。两个波函数之间的内积即为相应物理态之间交叠程度的量度,可应用于量子力学中最基本的波恩定律。该定律将跃迁几率与内积联系起来。薛定谔方程决定微观粒子体系的波函数随着时间将如何演化。该方程从数学上来说是一种波动方程,由此可以产生波粒二象性的概念。根据非相对论量子力学中波恩的统计解释,波函数的模方是一个实数,其可被解释为在给定位置处发现某微观粒子的几率密度。该模方具有有限值,且对于粒子所有自由度的求和等于1。由于波函数是复数,只有它的相对位相和相对数值才可被测得,它本身并不能独立地给出任何关于可观测量的数值或方向的有用信息。人们可以将量子力学算符(其本征值集合代表一组测量可能结果的集合)作用于波函数上,从而计算可观测量的统计分布。
对于多数同学来说,波函数概念比较抽象和晦涩。本文中,笔者结合自己的科研方向,针对需要学习量子力学课程的同学们简要介绍波函数在物理学稀土离子晶场参数从头计算中的重要应用。利用掺杂铈离子(Ce3+)体系计算实例的展示,期望同学们加强对波函数概念的理解,并激发他们对于量子力学课程学习的兴趣。需要指出的是,本文后续计算实例中的波函数采用矩阵表示形式。体系的波函数可以表示成一组正交归一完备基矢的线性展开,因此我们通常用该组展开系数(即为行或列矩阵)来表示该波函数。
2 基于波函数的稀土离子晶场参数的从头计算方法
Ce3+离子是稀土离子中最简单的情形(其基态只有1个4f电子),故本文以Ce3+离子掺杂体系为例来阐述波函数在稀土离子晶场参数从头计算中的重要应用。众所周知,在传统的稀土离子能级的参数化计算模型中,4fN态的有效哈密顿算符(Heff)主要由两部分组成,如下式所示:

其中,HAT为稀土离子4f电子的原子相互作用项,HCF为稀土离子4f电子与其配位环境之间的相互作用项(即晶场作用项)。HAT的具体形式如下:
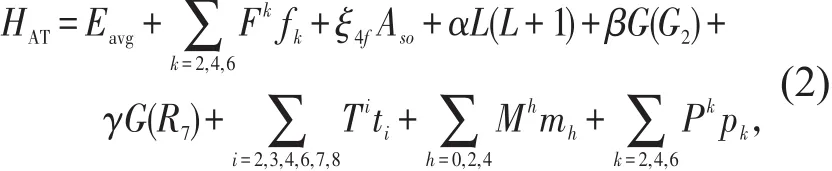
上式中各参量与相应算符的物理含义参见Carnall W T的论文[2],此处不再赘述。对于Ce3+离子的4f1组态,(2)式给出的HAT形式最为简单,仅包含旋轨耦合相互作用项,如下式所示:

其中,ξ4f为旋轨耦合作用参数(可以由经验拟合方法获得),ASO则为描述旋轨作用角度部分的算符。HCF的具体形式为

其中,Bkq表示Ce3+的4f电子与其配位体间物理作用的晶场参数,C(qk)为球谐张量算符。对于稀土离子的4fN组态,(4)式求和中k的取值为2,4和6,而q(整数)的绝对值不大于k。非零的q值则由晶体中掺杂Ce3+离子所处格位的点群对称性来决定。类似地,也可给出稀土离子4fN-15d能级参数化计算模型。该组态的Heff算符可由4fN-1组态、5d1组态的有效哈密顿算符以及两者之间的库伦相互作用哈密顿算符构成。对于Ce3+离子的5d1组态(由唯一的电子从4f基态激发所得到),它的有效哈密顿算符形式最为简单,因为它不含4fN-1态的Heff以及4fN-1与5d1组态之间的库伦作用项。
“网络是我们在全世界需要盟友和合作伙伴的地方。”然而,数据访问和共享可能打破个人与政府或企业原本建立的信任关系,由于大数据时代的元数据比以往任何时候都更容易创建,并且与其他数据形成聚合,突破隐私规则的束缚变得轻而易举,从而使人们丧失对隐私规则的预期。
一般来说,晶体中所掺杂稀土离子的晶场参量的取值可以通过拟合实验获得的光谱结构数据来确定。然而,有些稀土发光材料的实验能级数据缺失,相应的晶场参数的拟合确定就变得十分困难。我们提出的晶场参数从头计算方法并不依赖于实验光谱能级数据,而是基于最根本的晶体几何结构数据直接进行计算。针对Ce3+离子掺杂的发光材料,利用密度泛函理论(DFT)构型优化运算,得到该体系最稳定的超胞构型[3]。基于优化的超胞,构造镶嵌于基质晶格中的缺陷团簇,它是以Ce3+离子为中心并包含其最近邻一层或几层配位离子。然后对该镶嵌团簇模型进行量子化学从头计算以获得晶体中所掺杂Ce3+离子的能级和相应的波函数信息[4]。其中,从头计算采取的是完全活性空间自洽场(CASSCF)方法。
选取7个以Ce原子为中心的4f基函数(或5个以Ce原子为中心的5d基函数)来构造Ce的4 f(或5d)“模空间”。将量子化学从头计算获得的多电子波函数(CASSCF波函数)逐步投影到该模空间,获得Ce3+离子7个4f1态(或5个5d1态)的本征值以及相应本征矢所对应的矩阵,进而构造Ce3+离子4f1组态(或5d1组态)的有效哈密顿算符:

其中,Ep是Ce3+离子4f1(5d1)组态的能级矩阵(具有对角化形式),Vk则按照下式构造:

其中,Vp为量子化学从头计算输出的体系波函数通过逐步展开投影到4f(或5d)模空间的系数矩阵。
所选取体系的CASSCF波函数是组态函数(Slater行列式)的线性组合。值得注意的是,Slater行列式是一种符合电子交换反对称性要求的多电子波函数的描述方式。组态函数则由分子自旋轨道(具有分子轨道和自旋函数的乘积形式)产生。上述分子轨道按照以Ce原子为中心的基函数展开。至此,就可以基于量子化学从头计算方法构造Ce3+离子掺杂材料体系的有效哈密顿量算符。结合(1)式和(5)式,有效哈密顿量表达式中的各种相互作用参数(特别是我们所关心的晶场参数)就可以直接计算出来。
3 计算实例
选取Y3Al5O12:Ce3+(YAG:Ce3+)作为模型体系来展示上述计算方法。YAG:Ce3+是目前白光发光二极管(LED)商业化制作中被广泛应用的荧光材料。所掺杂Ce3+离子将占据YAG基质中8配位Y3+格位。该晶格格位的点群对称性为D2。因此,YAG中Ce3+离子非零4f和5d晶场参数的个数分别为9和5。基于镶嵌团簇从头计算获得的Ce3+离子4f1和5d1组态的能量本征值(单位为a.u.)如下式所示。

上述能量本征值直接取自CASSCF计算输出结果。假如以Ce3+离子最低的4f能级作为能量零点,则上述4f1组态能量本征值的相对值(单位为cm-1)为0,157,336,489,582,590和2 402;上述5d1组态能量本征值的相对值(单位为cm-1)为24 706,30 455,48 950,49 984和52 282。本文计算所获得的5d1能级值与Gracia J等人[5]的理论值符合得较好,但与实验测量的YAG:Ce3+激发光谱结果仍有一定偏差[6]。这可能是由于实际YAG:Ce3+材料中会存在各种缺陷或杂质,从而会影响Ce3+离子5d1能级结构。本文中所采用的镶嵌团簇模型则未考虑Ce3+局域配位环境中存在缺陷或杂质的情形。需要说明的是,上述能级计算值与实验测量结果之间的误差并不影响本文中关于稀土离子晶场参数从头计算方法的展示。
量子化学从头计算输出的YAG:Ce3+体系CASSCF波函数信息参见下列矩阵A和B((9)式和(10)式)。其中,矩阵A为该体系CASSCF多电子波函数(12个,即所选取的以Ce的4f和5d原子轨道为主要成分的体系多电子轨道)对于分子轨道(13个)的线性展开系数(12×13维,即每一行代表体系的一个多电子轨道)。矩阵B为13个分子轨道对于Ce的7个4f和5个5d原子轨道(Tesseral harmonic函数,田谐函数)的线性展开系数(13×12维,即每一行代表一个分子轨道)。

矩阵A和B直接取自从头计算的输出结果。我们需要对上述波函数结果进行变换,将其投影到4f和5d模空间。设矩阵C,令其等于矩阵A乘以B。因此,矩阵C为该体系多电子轨道对于Ce的4f和5d原子轨道的线性展开系数矩阵,其中每一列代表体系的一个多电子轨道。矩阵C的任意一列中的矩阵元对应4f和5d原子轨道的顺序依次为4f3+,4f2+,4f1+,4f0,4f1-,4f2-,4f3-,5d2+,5d1+,5d0,5d1-和5d2(-即田谐函数和)。进一步,通过下列公式将矩阵C的基函数由田谐函数变换为球谐函数(即Yqk函数),即获得新的系数矩阵(标记为矩阵D)。田谐和球谐函数的具体表达式参见Göfller-Walrand C等人的论文[7],

矩阵D中任意一行的12个矩阵元分别对应Ce原子轨道Y33,Y23,Y13,Y13,Y-13,Y-32,Y-23,Y22,,Y02,Y-12和Y-22(即球谐函数)的系数,而它的每一列对应一个本征矢量。上述获得的系数矩阵D即为前面所提到的Vp。至此,YAG:Ce3+体系的多电子波函数就通过逐步展开的方式投影到Ce的4f和5d模空间。按照前述方法,分别构造此体系4f和5d组态的有效哈密顿算符,并与唯象模型分析方法中所构造的有效哈密顿算符联系起来,从而求解出该体系中所掺杂Ce3+离子的晶场参数,如表1所示。从该表可以看出,基于量子化学从头计算获得非零4f和5d晶场参数的数目分别为9和5。这与根据Ce3+所占据格位的点群对称性(D2)预期的结果一致(不仅体现在参数数目上而且在具体非零项上)。
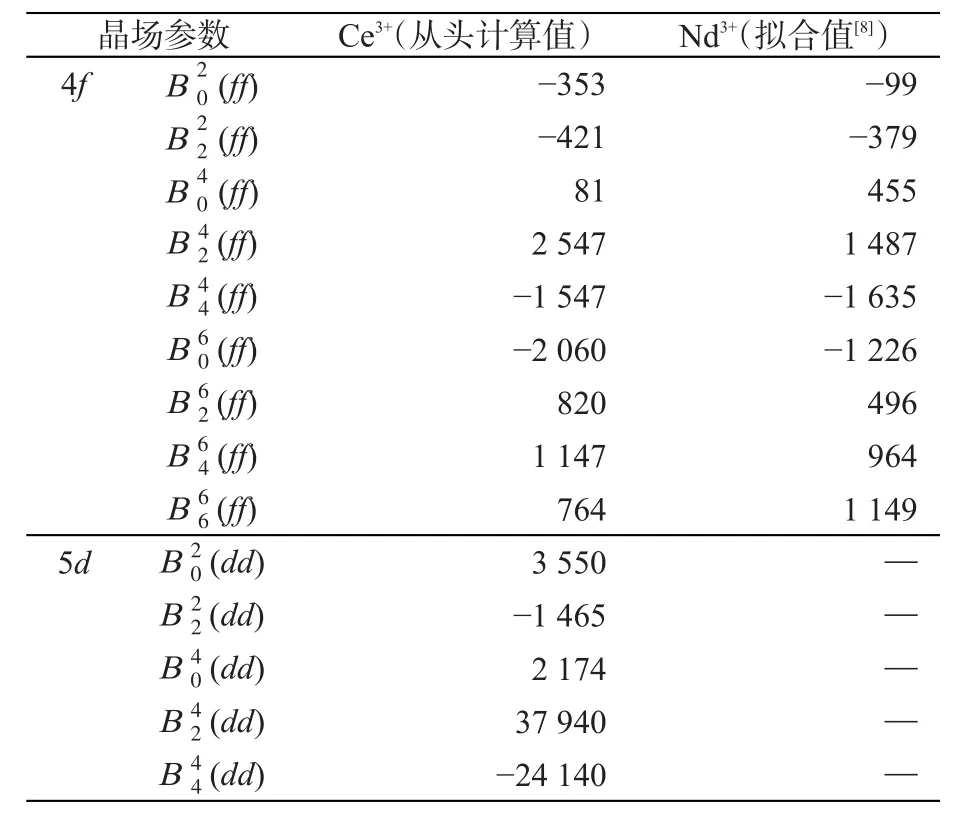
表1 从头计算获得的YAG:Ce3+晶场参数以及拟合获得的YAG:Nd3+晶场参数。单位:cm-1。
此外,根据Morrison C A等人的论文[8],YAG体系中所掺杂稀土离子一共有6套晶场参数。通过对比,从头计算获得的参数与他们给出的其中一套参数的符号完全一致。数值上的差别则是由于他们拟合获得的是Nd3+参数。上述结果说明了本文中所提出的基于波函数的稀土离子晶场参数从头计算方法的准确性。
4 结论
本文以YAG:Ce3+荧光粉体系为例,介绍了一种基于波函数的稀土离子晶场参数的从头计算方法,期望能够让量子力学初学者加强对波函数概念的理解。该方法主要利用量子化学从头计算输出的YAG中所掺杂Ce3+离子的4f和5d能级以及波函数结果来构造体系有效哈密顿量算符,并结合稀土离子能级参数化模型,在不依赖于实验能级数据的情况下准确计算出晶体中所掺杂稀土离子的晶场参数。
参考文献:
[1]曾谨言.量子力学[M].北京:科学出版社,2004:25-28.
[2]CARNALL W T,GOODMAN G L,RRJNAK K,et al.A systematic analysis of the spectra of the lanthanides doped into single-crystal LaF3[J].Journal of Chemical Physics,1989,90:3443-3457.
[3]WEN J,NING L,DUAN C,et al.A theoretical study on the structural and energy spectral properties of Ce3+ions doped in various fluoride compounds[J].Journal of Physical Chemistry C,2012,116(38):20513-20521.
[4]WEN J,YEUNG Y,NING L,et al.Thermal stabilities,electronic structures and optical properties of intrinsic defects and dopant cerium in Ca4F2Si2O7[J].Journal of Alloys and Compounds,2017,713:28-37.
[5]GRACIA J,SEIJO L,BARANDIARAN Z,et al.Ab initio calculations on the local structure and the 4f-5d absorption and emission spectra of Ce3+-doped YAG[J].Journal of Luminescence,2008,128(8):1248-1254.
[6]BLASSE G,BIRLA.Investigation of some Ce3+-activated phosphors[J].Journal of Chemical Physics,1967,47:5139.
[7]GSCHNEIDNER K A,EYRING L.Handbook on the physics and chemistry of rare earths(Vol.23)[M].Amsterdam:Elsevier Science B V,1996:121-283.
[8]GSCHNEIDNER K A,EYRING L.Handbook on the physics and chemistry of rare earths(Vol.5)[M].Amsterdam:North-Holland Publishing Company,1982:632.
