Autophagy plays a protective role in advanced glycation end productsinduced apoptosis of chondrocytes via regulation of tumor necrosis factor-α, nuclear factor-κB and reactive oxygen species
2018-05-09ZhiJiangSunYaYiXia
Zhi-Jiang Sun, Ya-Yi Xia
Orthopaedics Key Laboratory of Gansu Province, The Second Hospital of Lanzhou University, Lanzhou 730030, China.
1. Introduction
Advanced glycation end products (AGEs) are formedin vivoby metal-catalyzed glucose auto-oxidation and lipid peroxidation. A reducing sugar reacts with a protein (free amino groups in lysine,arginine or hydroxylysine residues) to form a labile, subsequently stabilized product, which produces an irreversible, non-enzymatic post-translational modification[1,2]. Once these are formed,AGEs are only degraded in the case of protein degradation that is prone to AGE formation[3]. The accumulation of AGEs affects the extracellular and intracellular structure and function in many different tissues and cell types. The most massive accumulation of AGEs will produce in tissues with low turnover, for instance,crystalline in the lens and collagen in the extracellular matrix of connective tissues, e.g., amyloid plaques[4,5], skin[6], tendon[7],bone and cartilage[8,9].
AGEs accumulation affects the function of tissues, contributing to the pathogenesis of diseases. Recent studies found that AGEs play a significant role in the development of age-related diseases, such as osteoarthritis[10,11]. Articular cartilages are prone to accumulate AGEs contributing to their low turnover. The accumulated AGEs increase the brittleness and stiffness of articular cartilage by reducing the synthesis of proteoglycan and collagen in the chondrocytes[11].As such, AGEs accumulation adversely affects the mechanical properties of the matrix driving the development of osteoarthritis.
Autophagy refers to the cellular degradative pathway that involves on turnover of cell constituents and serves as a short-lived survival mechanism under starvation environment by clearing away unfolded protein. Autophagy occurs at the low level in virtually all cells to perform the homeostatic function. However, it is rapidly up-regulated when cells need to generate intracellular nutrients and energy under certain stress conditions, such as starvation, infection, oxidative stress, protein aggregate accumulation and another irritation[12]. It is reported that AGEs also can induce autophagy in vascular smooth muscle cells and endothelial cells[13,14]. Generally, autophagy can promote cell survival by blocking apoptosis. However, autophagy also can result in cell death under certain conditions[15].
This study examined the effect of AGEs on cell viability,TNF-α andNF-κBexpression, ROS accumulation and apoptosis in human chondrocytes. Meanwhile, the role of autophagy on AGEs-induced adverse effect was investigated.
2. Materials and methods
2.1. Samples collection and cell culture
Cartilages were harvested from osteoarthritis patients with surgical procedures of total knee replacement (n=5, three males and two females, age range: 49-56 years old). The harvested cartilage was stored with amicrobic physiological saline. The cartilage was cut into 5-10 mm3slices after removing the subchondral lamina.Consequently, the complete treatment procedures were followed.Trypsin solution was used to soak the cartilage slices at room temperature for 30 min and then washed with phosphate buffer saline(PBS), followed by type II collagenase solution treatment at 37 ℃for 10 h. The isolated cells were maintained at 37 ℃ with 5% CO2in DMEM/F12 supplemented with 10% fetal bovine serum (HyClone,Logan, Utah, USA). The above procedures were performed under sterile conditions. Phase contrast images of the live chondrocytes were obtained using Olympus microscope.
2.2. AGEs treatment
AGE-BSA full-length protein was purchased from Abcam Company (ab51995). When the confluence of chondrocytes reached 80%, 100 μg/mL AGE-BSA was administered in the medium during the culture.
2.3. Cell viability assay
After AGEs treatment for 12, 24 and 48 h, the viability of chondrocyte populations in culture was quantified by 3-[4,5-dimethylthiazol2-yl]-2,5-diphenyl tetrazolium bromide (MTT).Cells were seeded at 3×103cells/well in a 96-well microplate in culture media with 10% FBS. MTT solution (0.5 mg/mL in PBS)was put into each well and then incubated at 37 ℃ for 4 h. Dimethyl sulfoxide (DMSO) was added after removing the supernatant. The absorbance was quantified using a microplate reader at 490 nm.
2.4. Rapamycin treatment induced autophagy
Autophagy inductor rapamycin (1 mmol/L) (Sigma, Co., St. Louis,MO, USA) was pretreated 2 h before AGEs incubation. MTT was used to measure cell viability at 12, 24, and 48 h after AGEs treatment. The time points for the experiments ofTNF-α andNF-κBexpression, intracellular ROS level and apoptosis were decided based on the result of MTT assay.
2.5. Western blot analysis
The chondrocytes were lysed using radio immunoprecipitation lysis buffer to extract protein. The protein in cell lysates was quantified using a spectrophotometer. The western blot analyses were performed using the rabbit monoclonal microtubule-associated protein 1 light chain 3 antibody (1:1 000 dilution; Cell Signaling Technology, Boston, MA, USA) and β-actin antibody (1: 3 000 dilution; Wuhan Sanying, Hubei, China), then labeled by horseradish peroxidase-conjugated antibody. The binds were visualized using enhanced chemiluminescent. Immunoblot signals were quantified using Gel-Pro Analyzer 4.0.
2.6. Expression of cellular TNF-α and NF-κB using quantitative real-time polymerase chain reaction (qRTPCR)
The expression ofTNF-α andNF-κBwere evaluated using qRT-PCR. Total RNA of chondrocytes was extracted using Trizol regent (Sigma, Co., St.Louis, MO, USA) and reverse-transcribed into cDNA according to instruction manual of an iScriptTMcDNA Synthesis Kit. The mRNA expression was quantified by two step SYBR Green RT-PCR. Relative fold change of gene was calculated using the comparative Ctequation. The relative amount of transcript was normalized against glyceraldehyde phosphate dehydrogenase(GAPDH) transcript. The amplification was in the presence of the following specific primer sets: 5´-CCT CAT CTA CTC CCA GGT-3´ and 5´-TAG ATG GGC TCA TAC CAG-3´ forTNF-α, 5´-TGG TGG AGG ATT TGC TGA GG-3´ and 5´-CCG TTG GGG TGG TCA AGA AG-3´ forNF-κB, 5´-CGG AGT CAA CGG ATT TGG TCG TAT-3´ and 5´-AGC CTT CTC CAT GGT GGT GAA GAC-3´ forGAPDH.
2.7. Measurement of intracellular ROS
Intracellular ROS level was measured using Carboxy-2’,5’-dichlorofluorescein diacetate (Carboxy-H2DCFDA) molecular probe(Invitrogen, Spartak Calder, CF, USA). After treatment, cells were incubated in medium containing 1 μmol/L DCFDA for 30 min at 37℃ in the dark. Cells were washed with PBS twice and dissociated enzymatically with trypsin added EDTA. Then cells were harvested by centrifugation and re-suspended in 500 μL PBS. Fluorescence absorbance was detected using a microplate reader (FCM, Thermo Fisher Scientific, Boston, MA, USA) with 488 nm/535 nm(excitation/emission wavelength).
2.8. Apoptosis analysis
The apoptosis rate was measured by flow cytometer (FCM, Thermo Fisher Scientific, Boston, MA, USA) using the Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (Nanjing Jiancheng,Jiangsu, China) according to the manufacturer’s instruction. Briefly,the chondrocytes were harvested by enzymolysis and centrifugation,re-suspended in binding buffer and added into FCM tube. Annexin V-FITC and PI were added in cells-suspension and incubated in the dark for 15 min before FCM measurement.
2.9. Statistical analysis
Statistical analysis was performed using Statistical Package for the Social Sciences (SPSS, Inc.). Statistically significant differences of means between three groups were determined by one-way ANOVA followed by the Post-Hoc test. The results are showed as mean ±standard deviation (Mean ± SD).P<0.05 was considered significant.
3. Results
3.1. Effects of AGEs and autophagy amelioration on cell viability
MTT assay showed that chondrocytes viability was remarkably decreased to 91.91% after 12 h incubation with AGEs (P<0.001),to 85.69% after 24 h incubation (P<0.001), and to 80.06% after 48 h incubation (P<0.001). At the same time point, the chondrocytes viabilities were 94.44%, 86.98% and 79.94% when the rapamycin was pretreated before AGEs incubation; It was significant only at 12 h (P=0.010) which suggested the response time to rapamycin.The chondrocytes viability under AGEs treatment or AGEs-added rapamycin treatment is shown Figure 1. The following results were that of experiments for chondrocytes treated with AGEs for 12 h.

Figure 1. Effect of advanced glycation end products (AGEs) on chondrocytes viability.
3.2. LC3 expression after rapamycin indicating autophagy
Rapamycin was administered to chondrocytes to induce autophagy.LC3 involved in formation of autophagosomal vacuoles, especially LC3-II is regarded as the biomarker of autophagy. Hence, the expression of cellular LC3 protein was measured using western blot to present the level of autophagy. Compared to that with no treatment, the total LC3 (including LC3-I and LC3-II) expression of chondrocytes was significantly increased both with AGEs treatment(P=0.015) and with AGEs-added rapamycin treatment (P<0.001)(Figure 2). The densitometry of LC3-II/β-actin in chondrocytes treated with AGEs added rapamycin was much higher than that in chondrocytes without treatment and AGEs treatment (bothP<0.001).There was no significance between chondrocytes with AGEs treatment and chondrocytes without treatment (Figure 2).

Figure 2. Densitometric analysis of cellular light chain (LC3) protein.In the bar, n = 5 in each group. *P<0.05, **P<0.001.
3.3. AGEs increased the expression of TNF-α and NF-κB of chondrocytes and autophagy receded the change
The effects of AGEs and autophagy on the expression ofTNF-α andNF-κBin chondrocytes were measured by qRT-PCR.Compared to that without treatment, the RNA expression levels ofTNF-α andNF-κBin chondrocytes with AGEs incubation were significantly higher (2.46-fold and 2.16-fold, bothP<0.001). In the case of AGEs-added rapamycin treatment, the RNA expression levels ofTNF-α andNF-κBin chondrocytes were reduced compared to that with single AGEs incubation. However, they were also higher than those without treatment (1.75-fold and 1.62-fold,P=0.004 andP<0.001, respectively).
3.4. AGEs increased intracellular ROS accumulation and autophagy reversed the change
To test the effect of AGEs on the cellular ROS levels, cells were stained with DCFDA and detected by FCM. The levels of cellular ROS increased when chondrocytes were with AGEs incubation for 12 h (P<0.001). The fluorescence absorbance of cellular ROS production was 284.4-334.4. When the chondrocytes were pretreated with rapamycin before AGEs incubation, the average fluorescence absorbance of cellular ROS production was 308.4, which was lower than that with AGEs incubation (P=0.012) but higher than that with no treatment (P=0.007).
3.5. AGEs accelerated chondrocytes apoptosis and autophagy suspended apoptosis
To evaluate the apoptosis of chondrocytes under different conditions, the chondrocytes were stained by Annexin V-FITC/PI and detected by FCM (Figure 3). AGEs accelerated chondrocytes apoptosis, and the rates of apoptosis were from 6.41% (95%CI:4.32%-8.50%) to 24.31% (95%CI: 18.00%-30.63%). With the rapamycin pretreatment, the rate of apoptosis returned to 15.18%(95%CI: 13.48%-16.88%) under the autophagy protection.
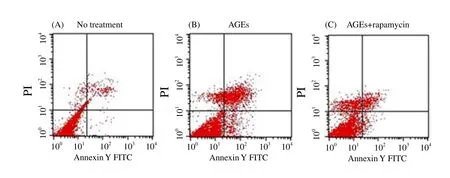
Figure 3. Chondrocytes apoptosis after culture with advanced glycation end products (AGEs) treatment and AGEs-added rapamycin treatment.
4. Discussion
AGEs are lipids or proteins which turn glycated after sugars exposure. AGEs accumulation plays a pivotal role in osteoarthritis which is predisposed by aging[10,16,17]. Accumulation of AGEs decreases collagen synthesis and turnover, increases matrix metalloproteinase (MMP)-1, MMP-3, and MMP-13 in human osteoarthritic chondrocytes[18], and alters the tensile properties of articular cartilage[19]. Our results indicate that AGEs decreased cell viability, increased TNF-α and NF-κB expression, upregulated ROS production and induced apoptosis in human articular chondrocytes; however, autophagy significantly reversed AGEsinduced above damages.
AGEs-bound AGEs receptor stimulates signaling pathways linked to pro-inflammatory, activating various inflammatory genes and many signaling cascades[20,21]. Some researchers have showed that TNF-α is in the upstream in the signaling cascades. In human umbilical vein endothelial cells, the evoked sequences by TNF-α include the following events: NADPH oxidase stimulation to ROS generation to mitochondrial respiratory chain activation to NF-κB activity stimulation to RAGE expression induction[22]. But some researches showed that TNF-α is in the downstream of AGEsbound AGEs receptor and NF-κB[23]. In this study, although we cannot identify the relationship of sequenced activation between TNF-α and NF-κB, we verified AGEs increased TNF-α and NF-κB expression in human chondrocytes.
AGEs play a crucial role in the oxidative stress injury and the apoptosis[24]. In the endothelium, AGEs block nitric oxide activity and induce mitochondrial dysfunction thus cause the ROS production, which facilitates the production of mitochondrial superoxide afterward[25,26]. The mitochondrial dysfunction and high levels of ROS cause apoptosis through caspase activation.The results of this study also showed AGEs increased the levels of ROS accumulation and the apoptosis in chondrocytes. AGEsinduced ROS generation is partly through NF-κB activation in human aortic endothelial cells[27]. Additionally, as mentioned above,ROS generation also can induce NF-κB activity. TNF-α can activate both apoptotic and survival signals mediating apoptosis,proliferation, differentiation, and survival of cells[28]. Tumor necrosis factor (TNF) activates apoptosis and anti-apoptosis pathways simultaneously. TNF binding to TNF receptor 1 sequentially recruits TNF receptor-associated death domain, Fas-associated death domain, Fas-associated death domain-like interleukin-1β converting enzyme, and caspase-3, leading to apoptosis[29]. However, both effects of TNF are mediated through the production of reactive oxygen intermediates. There may be a cross talking among TNF-α,NF-κB, ROS generation and apoptosis under AGEs provoking.
Autophagy is a protection mechanism that involves in the homeostasis maintenance of cells in reaction to many forms of stress such as oxygen, nutrient, chemotherapeutics and growth factor deprivation. The relationship between ROS and autophagy is mutual. ROS accumulation induces autophagy, in turn, autophagy serves to lower ROS level[30,31]. In general, autophagy is valuable to cell survival in a diverged condition; however, excessive autophagy can cause cell death. In this study, we showed that AGEs exerted their adverse effects on the cell viability, TNF-α and NF-κB expression, ROS accumulation and apoptosis. Meanwhile, when the chondrocytes were pretreated with the autophagy inductor rapamycin, all the adverse effects were improved. This indicated autophagy was a defense mechanism to protect cartilage damage when it is adversely attacked by AGEs against damaged organelles and harmful metabolites, delaying the ROS accumulation and reducing apoptosis.
In conclusion, this study confirms autophagy plays a protective role in AGEs-induced apoptosis of chondrocytes possibly via regulation of TNF-α, NF-κB and ROS. However, the potential molecular mechanisms are still elusive to a large extent. The future studies should focus on the important question. The results also suggest that the novel pharmacological actions against AGEs-stimulated oxidative stress and apoptosis of chondrocytes are a potential measure for treating osteoarthritis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
[1] Singh R, Barden A, Mori T, Beilin L.Advanced glycation end-products:A review.Diabetologia2001; 44: 129-146.
[2] Sugimoto K, Yasujima M, Yagihashi S. Role of advanced glycation end products in diabetic neuropathy.Curr Pharm Des2008; 14: 953-961.
[3] Lai KN, Leung JCK, Chan LYY, Li FFK, Tang SCW, Lam MF, et al.Differential expression of receptors for advanced glycation end-products in peritoneal mesothelial cells exposed to glucose degradation products.Clin Exp Immunol2004; 138: 466-475.
[4] Ko SY, Lin YP, Lin YS, Chang SS. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species.Free Radic Biol Med2010; 49: 474-480.
[5] Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, et al. Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature.Hypertension2012;60: 188-197.
[6] Mulder DJ, van Haelst PL, Gross S, de Leeuw K, Bijzet J, Graaff R, et al.Skin autofluorescence is elevated in patients with stable coronary artery disease and is associated with serum levels of neopterin and the soluble receptor for advanced glycation end products.Atherosclerosis2008; 197:217-223.
[7] Li Y, Fessel G, Georgiadis M, Snedeker JG. Advanced glycation endproducts diminish tendon collagen fiber sliding.Matrix Biol2013; 32:169-177.
[8] Kume S, Kato S, Yamagishi SI, Inagaki Y, Ueda S, Arima N, et al.Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage,and bone.J Bone Miner Res2005; 20: 1647-1658.
[9] Ganeko K, Masaki C, Shibata Y, Mukaibo T, Kondo Y, Nakamoto T,et al. Bone aging by advanced glycation end products: A multiscale mechanical analysis.J Dent Res2015; 94: 1684-1690.
[10] DeGroot J, Verzijl N, Wenting-van Wijk MJG, Jacobs KMG, Van El B, Van Roermund PM, et al. Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis.Arthritis Rheumatol2004; 50: 1207-1215.
[11] Serrao PRMS, Vasilceac FA, Gramani-Say K, Lessi GC, Reiff RBM,Mattiello-Sverzut AC, et al. Expression of receptors of advanced glycation end product (RAGE) and types I, III and IV collagen in the vastus lateralis muscle of men in early stages of knee osteoarthritis.Connect Tissue Res2014; 55: 331-338.
[12] Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion.Nature2008; 451: 1069-1075.
[13] Xie Y, You SJ, Zhang YL, Han Q, Cao YJ, Xu XS, et al. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells.Mol Med Rep2011; 4: 459-464.
[14] Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells.Int J Mol Med2012; 29: 613-618.
[15] Kroemer G, Levine B. Autophagic cell death: The story of a misnomer.Nat Rev Mol Cell Biol2008; 9: 1004-1010.
[16] Li Y, Wei X, Zhou J, Wei L. The age-related changes in cartilage and osteoarthritis.Biomed Res Int2013: 2013: 916530.
[17] Yang X, Gandhi C, Rahman MDM, Appleford M, Sun LW, Wang X.Age-related effects of advanced glycation end products (ages) in bone matrix on osteoclastic resorption.Calcif Tissue Int2015; 97: 592-601.
[18] Nah SS, Choi IY, Yoo B, Kim YG, Moon HB, Lee CK. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13,and TNF-alpha in human osteoarthritic chondrocytes.FEBS Lett2007;581: 1928-1932.
[19] Chen AC, Temple MM, Ng DM, Verzijl N, DeGroot J, TeKoppele JM,et al. Induction of advanced glycation end products and alterations of the tensile properties of articular cartilage.Arthritis Rheumatol2002; 46:3212-3217.
[20] Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B,et al. Understanding RAGE, the receptor for advanced glycation end products.J Mol Med2005; 83: 876-886.
[21] Prasad K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: myth or reality.Int J Angiol2014; 23: 11-16.
[22] Mukherjee TK, Mukhopadhyay S, Hoidal JR. The role of reactive oxygen species in TNF alpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells.Biochim Biophys Acta2005; 1744: 213-223.
[23] Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products - A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis.Circ Res1999;84: 489-497.
[24] Alikhani ZB, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of proapoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways.J Biol Chem2005; 280: 12087-12095.
[25] Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE,Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes.J Am Soc Nephrol2009; 20: 742-752.
[26] Wu Q, Zhong ZM, Zhu SY, Liao CR, Pan Y, Zeng JH, et al. Advanced oxidation protein products induce chondrocyte apoptosis via receptor for advanced glycation end products-mediated, redox-dependent intrinsic apoptosis pathway.Apoptosis2016; 21: 36-50.
[27] Morita M, Yano S, Yamaguchi T, Sugimoto T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-kappa B activation in human aortic endothelial cells.J Diabetes Complications2013; 27: 11-15.
[28] Royuela M, Rodriguez-Berriguete G, Fraile B, Paniagua R. TNF-alpha/IL-1/NF-kappa B transduction pathway in human cancer prostate.Histol Histopathol2008; 23: 1279-1290.
[29] Zhang X, Vallabhaneni R, Loughran PA, Shapiro R, Yin XM, Yuan Y, et al. Changes in FADD levels, distribution, and phosphorylation in TNF alpha-induced apoptosis in hepatocytes is caspase-3, caspase-8 and BID dependent.Apoptosis2008; 13: 983-992.
[30] Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS:Physiology and pathology.Trends Biochem Sci2011; 36: 30-38.
[31] Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy.Cell Death Differ2009; 16: 1040-1052.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- An updated systematic review of Zika virus-linked complications
- Biological, chemical and pharmacological aspects of Madhuca longifolia
- Scenario of dengue infection & its control in Pakistan: An up-date and way forward
- Cytotoxic, kinetics of inhibition of carbohydrate-hydrolysing enzymes and oxidative stress mitigation by flavonoids roots extract of Dicoma anomala (Sond.)
- Evaluation of antiparasitic, anticancer, antimicrobial and hypoglycemic properties of organic extracts from Panamanian mangrove plants
- Neuroprotection by misoprostol against rotenone-induced neurotoxicity in rat brain