采用复合锂源制备的LiFePO4/C材料及其碳含量优化
2018-05-05周盈科
吴 关 周盈科
(武汉科技大学,省部共建耐火材料与冶金国家重点实验室,武汉 430081)
0 引 言
锂离子电池在电动工具市场、电动汽车(EV)市场和储能系统(ESS)中的应用日益普遍,从当前的发展趋势看,未来锂离子电池在人们的日常生活中将被更加广泛地应用[1-3]。迄今为止,锂离子电池的性能特征主要取决于所采用的正极材料[4-6]。锂离子电池的能量密度、功率密度与使用寿命,也主要受到正极材料动力学性能的制约。LiFePO4/C材料作为一种具有优秀循环性能、高安全性能与低成本的正极材料,已经较为成熟地商业化生产,并应用于电动工具、电动汽车与储能系统等领域。目前,固相合成法仍然是商业化应用最广泛的LiFePO4/C材料制备方法,这主要是由于固相合成法易于大规模复制生产,工业生产过程易于控制,生产成本较低廉[7-10]。
由于LiFePO4材料的本征电子电导率与锂离子扩散速率极低,需要在颗粒表面进行碳包覆以改善其动力学性能[11]。包覆碳层的存在能够提高材料的电导率,控制材料的颗粒尺寸,但是碳包覆量会影响材料的比表面积与加工性能[12-17]。因此优化碳包覆量是获得具有最优综合性能LiFePO4/C材料的必要研究课题[18-19]。本文以FePO4为前驱物,创新地采用Li2CO3与LiOH·H2O为复合锂源,通过过固相合成法制备了具有不同碳含量的LiFePO4/C材料,对所制备的LiFePO4/C样品进行了全面的物理性质、加工性能与电化学性能测试,最终确定了LiFePO4/C材料的最优碳包覆量。本文采用Li2CO3与LiOH·H2O为复合锂源制备LiFePO4/C材料,不同于商业化LiFePO4/C材料常用的固相合成法单纯使用Li2CO3为锂源。所制得的LiFePO4/C材料晶相结构完整、纯度高、无杂相,表现出优秀的加工性能与电化学性能。
1 实验方法
1.1 LiFePO4/C材料的制备
以 FePO4(≥99.5%(w/w))、Li2CO3(≥99.5%(w/w))、LiOH·H2O(≥99.5%(w/w))与葡萄糖(≥99.5%(w/w))为原料,所采用的 FePO4原料中 nFe∶nP=0.97∶1,复合锂源由 LiOH·H2O与 Li2CO3按照质量比 2∶8混合而成,通过调节复合锂源的用量使 nLi∶nFe∶nP=1.02∶0.97∶1。将FePO4、复合锂源、葡萄糖依次加入砂磨机中,以去离子水为溶剂,经过充分研磨后得到混合均匀的流态浆料,使用喷雾干燥器将浆料干燥后,在N2气氛下升温至720℃进行煅烧并保温10 h,最后经过气流粉碎机破碎得到LiFePO4/C材料。在制备过程中通过控制葡萄糖的添加量,控制最终所得LiFePO4/C材料的碳含量。本文共制备了4个具有不同碳含量的LiFePO4/C样品,设计碳含量的质量百分数分别为1%、2%、3%与4%,并分别将样品命名为 LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4。
1.2 LiFePO4/C样品表征
用Bruker D8型 X射线衍射仪(XRD)测试LiFePO4/C的晶型结构,使用波长λ为0.154 18 nm的Cu Kα射线为发射源,在40 kV的管电压和100 mA 的管电流,2θ=15°~85°的扫描范围内进行测试。使用TriStarⅡ3020型比表面积测试仪,以氮气吸附-脱附的方法测试样品的比表面积,在实验前将粉体在200℃下真空处理3 h以排除表面吸附物质对实验的影响,采用Brunauere-Emmette-Teller(BET)法计算确定样品的比表面积大小。使用Dekai HCS-140型碳硫分析仪测试所得LiFePO4/C样品中的碳含量,灼烧温度为1 200℃,采用红外光度法测算样品的碳含量。使用ZEISS Sigma-02-33型扫描电子显微镜(SEM)在10 kV工作电压下对所得样品进行测试。使用FEI F20型透射电镜(TEM)在10 kV工作电压下观测样品的微观形貌。
1.3 LiFePO4/C电化学性能测试
首先采用以单质锂作为对电极的模拟电池即半电池对所得样品进行电化学性能测试,扣式电池的充放电测试在2.0~3.8 V电压范围内进行,采用Land BTS-5V-50 mA电池测试系统。扣式电池的制作方法如下:首先称取质量比为90∶5∶5的LiFePO4/C、Super-P和PVDF,并充分搅拌形成浆料,将经过充分搅拌分散后的浆料涂覆于集流体铝箔上,随后将极片在85℃下干燥,并使用辊压机进行冷压获得均匀平整的极片。使用冲片机将极片制成直径为14 mm的圆片,并再次干燥。称量极片质量并计算活性物质质量后,将极片在使用高纯氩气保护的手套箱中组装成2430型扣式电池,正极极片中的活性材料质量为8.4~9.1 mg·cm-2。扣式电池中使用的负极为金属锂片。
对所制得的扣式电池样品在2.0~4.2 V电压范围内,使用Bio-Logic VMP3B型电化学工作站以0.1 mV·s-1的扫描速率进行循环伏安(CV)测试。采用ZAHNER Im6ex型电化学工作站以5 mV的外加交流信号幅值,在100 mHz~1 MHz扫描频率范围内对所制得的电极样品进行交流阻抗(EIS)测试。
为进一步确认所制得的样品在实际应用过程中的长期可靠性,还针对所得LiFePO4/C材料制作以商业化人造石墨为负极的额定容量为2 Ah的软包全电池并测试其电化学性能。软包全电池制作方法如下:称取质量比为 94∶2∶4 的 LiFePO4/C、Super-P和PVDF,并在NMP溶液中均匀分散,通过调节NMP的加入量将浆料的固含量控制在50%左右,黏度控制在5 000~8 000 mPa·s左右。再将搅拌均匀的浆料过筛后经过涂覆、干燥、冷压、分条得到活性物质为LiFePO4/C的阴极极片。随后将阴极极片、隔离膜、活性物质为石墨的阳极极片卷绕成裸电芯,使用铝塑膜进行塑封后,经过注液、老化、除气、再封装即得到待测的软包全电池。对所制得的软包全电池样品在2.0~3.65 V电压范围内,在1C倍率下,使用Neware 5V30A电池测试系统测试全电池的长期存储性能。
2 结果与讨论
2.1 物理特性测试
通过碳硫分析仪确认了4个样品的实际碳含量 ,LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 样 品 的实测碳含量分别为1.03%、1.95%、2.92%与4.11%(w/w),与设计值十分接近。通过氮气吸附-脱附测试仪测得 LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 材料的比表面积分别为 13、16、20 与 24 m2·g-1, 说明随着碳含量的增加,材料的比表面积也显著增加,这主要是由于包覆在LiFePO4颗粒表面的有机裂解碳具有很高的比表面积,LiFePO4/C的比表面积增加主要是由于表面包覆碳导致的。
2.2 XRD测试
图1为样品的XRD图,与标准卡片PDF No.83-2092对比可知,4个样品均能归属于橄榄石型结构LiFePO4[20]。所有样品的XRD图中均没有出现杂峰,说明具有不同碳含量的4个LiFePO4/C材料均显示出良好的晶型结构。尽管4个LiFePO4/C样品具有不同的碳含量,但在XRD图中均未发现碳的特征峰,这主要是由于葡萄糖高温裂解后产生的碳大部分都是以无定形的状态存在且总量较低。
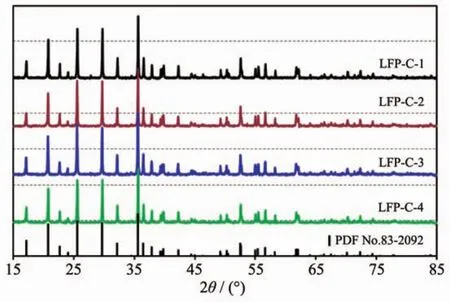
图1 四个LiFePO4/C样品的XRD图Fig.1 XRD patterns of as-prepared LiFePO4/C samples
2.3 SEM与TEM表征
图 2(a~d)分别为 LFP-C-1、LFP-C-2、LFP-C-3 与LFP-C-4材料的SEM图。材料的一次颗粒粒径随着碳含量的增加而减小,LFP-C-1材料由于碳含量仅为1.03%,样品中存在微米级的大颗粒。LFP-C-1与LFP-C-2样品中均未发现明显过剩的残留碳,说明LiFePO4/C材料中的碳含量低于2%时包覆效果较好,颗粒表面状态较稳定。当碳含量提高至3%左右时,LFP-C-3样品中可观测到明显的残余裂解碳分布在颗粒表面,碳含量提高至4%左右时,LFP-C-4样品中的裂解碳已经严重过剩,这些过剩的碳材料并未较好地包覆LiFePO4颗粒,反而直接导致了材料的比表面积显著增大,可能加剧材料与电解液之间的副反应,对循环与存储等电化学性能的发挥造成不利影响。因此,LiFePO4/C材料中的碳含量应尽量控制在3%以内,减少样品中多余的裂解碳,使材料具有较高导电能力的同时,尽可能避免与电解液发生副反应,平衡导电率与综合电性能[21-22]。

图2 (a)LFP-C-1,(b)LFP-C-2,(c)LFP-C-3和(d)LFPC-4的SEM图Fig.2 SEM images of(a)LFP-C-1,(b)LFP-C-2,(c)LFPC-3 and(d)LFP-C-4
为了进一步验证不同碳含量对表面包覆状态的影响,分别对4个样品进行了TEM表征(图3)。由于LFP-C-1材料的碳含量仅为1.03%,LiFePO4颗粒表面包覆的碳层厚度仅为1 nm左右,且出现了表面包覆不连续的现象(图3a),使得材料的导电能力得不到充分提高。碳含量为2%左右的LFP-C-2材料的表面包覆碳层厚度为3~5 nm,包覆效果连续且均匀,是包覆效果最佳的样品(图3b)。而当碳含量进一步提高时,LFP-C-3与LFP-C-4材料的颗粒表面均出现了明显的过剩碳材料(图3(c,d)),这些过剩的碳材料并未紧密地包覆于LiFePO4颗粒表面,不能达到显著提高材料导电性的目的,反而会增加副反应,恶化电化学性能。TEM与SEM表征结果一致,证明当碳含量在2%左右时,LiFePO4颗粒获得了最优的包覆效率与效果。

图3 (a)LFP-C-1,(b)LFP-C-2,(c)LFP-C-3,(d)LFP-C-4的TEM图Fig.3 TEM images of(a)LFP-C-1,(b)LFP-C-2,(c)LFPC-3 and(d)LFP-C-4
2.4 扣式电池倍率性能测试
图 4(a~d)分别为 LFP-C-1、LFP-C-2、LFP-C-3 与LFP-C-4样品的扣式电池倍率放电性能曲线。对比倍率放电曲线可知,LFP-C-1材料在大倍率放电时的电压平台最低,LFP-C-2次之,而LFP-C-3与LFPC-4材料在大倍率放电时均展现出较高的电压平台,说明碳含量仅为1.03%的LFP-C-1材料受限于动力学性能,在大倍率放电时表现出较大的极化,随着碳含量的增加,材料的导电性能得到提升,极化也随之减小。在大倍率放电时,4个样品的放电克容量也随着碳含量的增加而增加,如2C放电时,LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 的放电克容量分别为 104.0、128.3、138.7 与 138.8 mAh·g-1,5C放 电 时 ,LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 的放 电 克 容 量 分 别 为 77.2、105.7、126.8与 124.8 mAh·g-1,说明碳含量越高LiFePO4/C材料的大倍率放电能力越强,而当碳含量达到3%左右时,进一步提高碳含量已无法明显改善倍率性能。但是,在小倍率放电时,却出现了不同于大倍率放电时的规律,即碳含量为1.95%的LFP-C-2样品表现出了较高的放电克容量。在0.1C倍率下放电时,LFP-C-1、LFP-C-2、LFP-C-3与LFP-C-4的放电克容量分别为148.9、158.7、157.2 与 158.2 mAh·g-1, 在 0.2C 倍率放 电 时 ,LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 的放电克容量分别为 145.6、156.5、152.6与 153.7 mAh·g-1。结合前文的SEM形貌分析,这主要是由于当碳含量在2%左右时,LiFePO4颗粒获得了最优的包覆效率与效果,使得小倍率下LFP-C-2具有最高的克容量,但由于LFP-C-2的总碳含量只有1.95%,其实际导电能力低于碳含量分别为2.92%和4.11%的LFP-C-3和LFP-C-4,所以在大倍率下依然存在较大极化,克容量低于LFP-C-3与LFP-C-4。
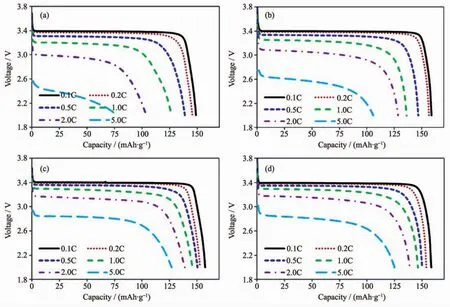
图4 (a)LFP-C-1,(b)LFP-C-2,(c)LFP-C-3,(d)LFP-C-4的倍率性能图Fig.4 Rate performances of(a)LFP-C-1,(b)LFP-C-2,(c)LFP-C-3 and(d)LFP-C-4
2.5 扣式电池循环测试
图5 为4个样品的0.5C充放电循环曲线对比图。4个样品的循环测试均在0.1C倍率下经过2次循环活化后进行,在进行了60次0.5C充放电循环后 ,LFP-C-1、LFP-C-2、LFP-C-3 与 LFP-C-4 材 料 的容量保持率分别为91.3%、99.0%、97.0%与96.4%。相较而言,LFP-C-1样品表现出的循环性能最差,这是因为LFP-C-1的碳包覆量仅为1.03%,无法完全包覆LiFePO4颗粒表面,使得LiFePO4颗粒本体与电解液直接接触发生副反应,反应产物使内阻增大,循环性能变差。LFP-C-3与LFP-C-4材料中存在多余的裂解碳,这部分裂解碳对提高材料的导电性能已无明显作用,却大大提高了材料的比表面积,为材料与电解液之间的副反应提供了大量的活性位点,因此循环性能也受到不利影响。而LFP-C-2材料碳包覆量适中,既保证了LiFePO4颗粒被碳层较完整地包覆,材料中又不存在多余的裂解碳,具有优秀的表面结构稳定性,因此表现出最优的循环性能。

图5 四个样品的循环性能曲线对比图Fig.5 Comparative cyclic curves of four samples
2.6 扣式电池动力学性能测试
对LiFePO4材料进行碳包覆的过程能够改善其电子导电性,且碳包覆能够抑制LiFePO4一次颗粒粒径的生长从而提高其锂离子扩散性[23-24],所以碳包覆量对材料的动力学性能有显著地影响。图6(a,b)分别为4个样品在25℃与0℃的EIS对比曲线,图7为4个样品的CV对比曲线。在25℃时,LFPC-1、LFP-C-2、LFP-C-3、LFP-C-4 的电荷转移阻抗分别为 395、142、86、48 Ω;在 0 ℃时,LFP-C-1、LFP-C-2、LFP-C-3、LFP-C-4 的电荷转移阻抗分别为 493、197、136、69 Ω。 碳含量越高的样品,其电化学阻抗越低,说明LiFePO4/C的碳包覆量是影响其动力学性能的关键因素,提高碳包覆量能够显著地降低材料的电化学阻抗。LFP-C-1材料在不同温度下的阻抗均远大于其他3个样品,其受温度的影响也非常明显,说明1.03%的碳包覆量不能够满足材料的性能需求。
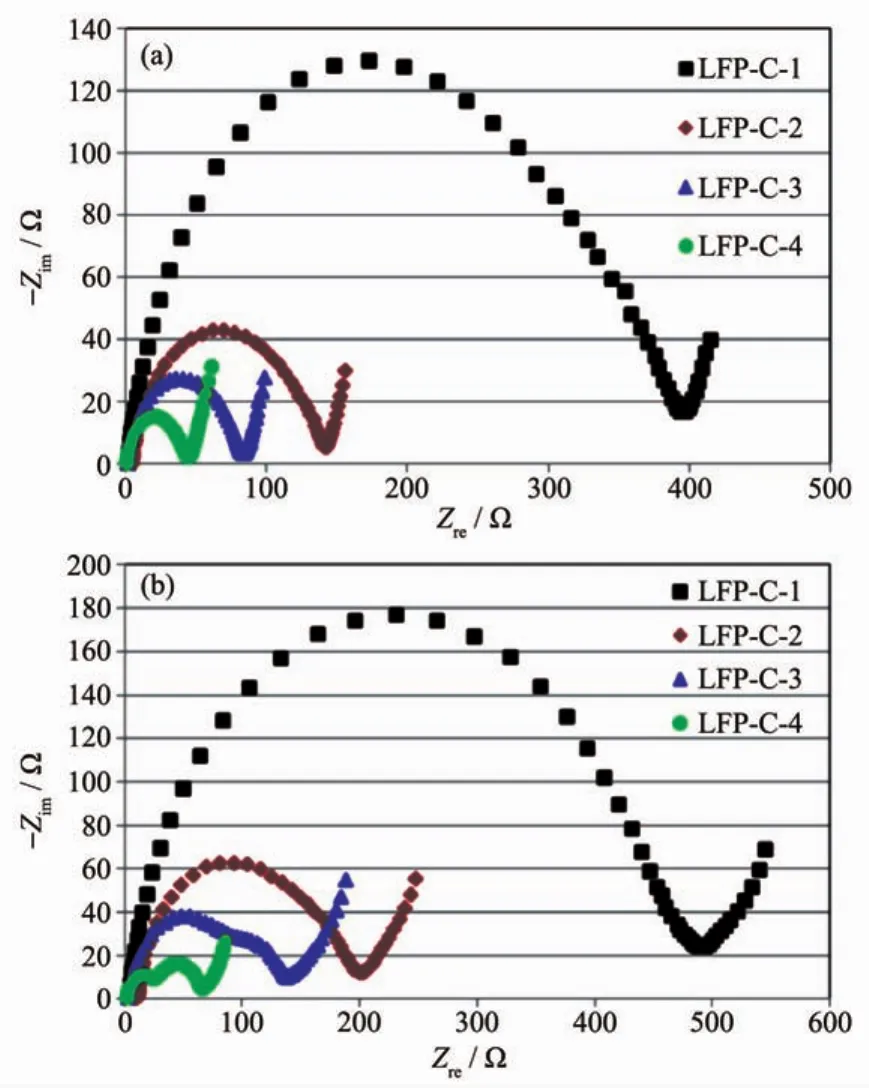
图6 在(a)25和(b)0℃下4个样品的EIS图谱Fig.6 EIS spetra of four samples at(a)25 and(b)0℃
由图7中可知,LFP-C-2、LFP-C-3与LFP-C-4三个样品的CV曲线略有差异,但整体处于同一水平内,LFP-C-1样品的氧化还原峰最宽泛,进一步表明了LFP-C-1材料的极化最大,LFP-C-3样品的氧化还原峰最尖锐电压差值最小,这主要是由于LFP-C-3样品的碳含量较高,导电性能优于LFP-C-2材料,而由残余裂解碳引发的副反应较LFP-C-4材料少。
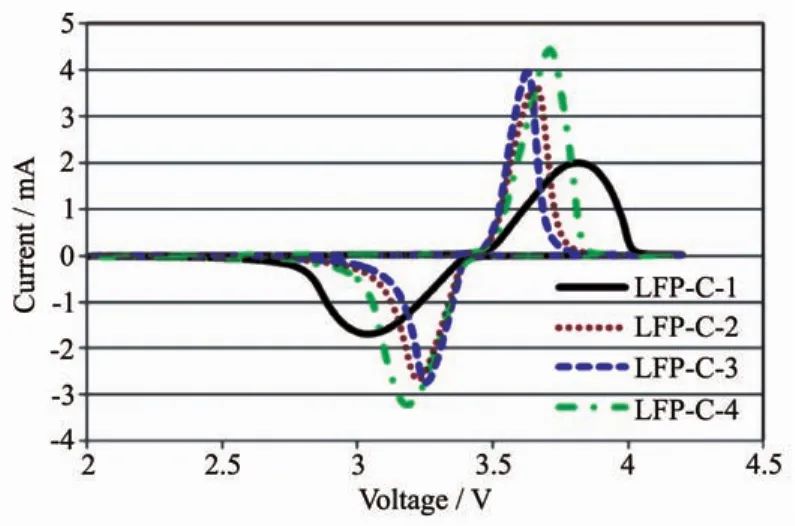
图7 四个样品的CV图Fig.7 CV curves of four samples
2.7 全电池长期存储性能测试
为了验证具有不同碳含量的LiFePO4/C材料在实际应用时的性能表现,本文制作了软包全电池对4个样品分别进行实效性能测试,所制作的全电池使用商品化的石墨和电解液进行电芯体系匹配,软包全电池的额定容量为2 000 mAh。
图8(a,b)分别为4个样品在25与60℃下的存储性能对比图,由测试结果可知,样品在常温与高温下的存储性能表现出一致的趋势,即存储性能与碳含量呈负相关,碳含量越高的材料存储性能越差[25-26]。由于LFP-C-1材料的碳含量仅为1.03%,比表面积也最小,其在存储过程中提供给电解液与材料发生表面副反应的活性位点最少,因此LFP-C-1材料在存储过程中损失的容量最少,表现出最优的存储性能。而LFP-C-4材料由于碳含量达到4.11%,材料中存在大量多余的无定形热裂解碳,这些碳材料的比表面积相当大,显著增加了材料与电解液的接触面积,为材料与电解液之间的副反应提供了活性位点与反应场所,材料与电解液之间发生副反应会消耗体系内部的活性锂,造成容量损失,因此LFP-C-4存储性能差,且对温度敏感,高温下存储后的容量保持率降幅更加明显。4个样品在存储过程中表现出不同容量衰减速率,碳含量为1.95%的LFP-C-2材料的容量衰减速率较LFP-C-1材料略有增加。LFP-C-3样品的碳含量达到2.92%,存储性能明显恶化。在60℃下存储78 d后,碳含量较高的LFP-C-3与LFP-C-4样品的容量保持率分别仅为87.1%与83.6%。结合存储测试与其他电化学性能测试结果可知,LiFePO4/C材料的包覆碳含量需要控制在一定范围内,以获得优秀的长期存储性能,同时兼顾其他电化学性能,采用本文介绍的固相合成法制备LiFePO4/C材料时,应将碳含量的上限控制在2%左右。
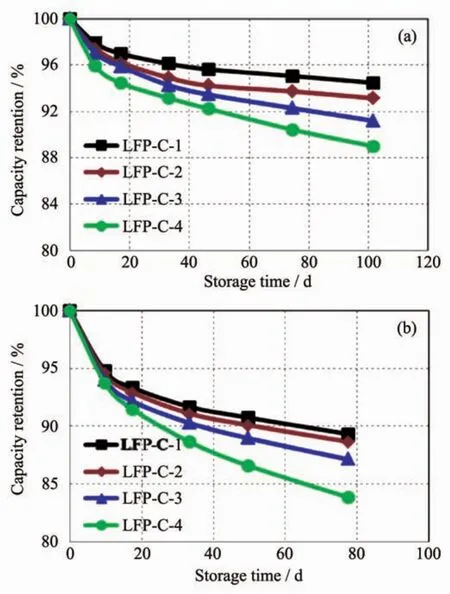
图8 四个样品在(a)25和(b)60℃下的全电池存储性能曲线Fig.8 Storage curves of four samples at(a)25 and(b)60℃
3 结 论
本文使用固相合成法,以FePO4为前驱物,LiOH·H2O与Li2CO3为复合锂源,葡萄糖为碳源,制备了4个具有不同碳含量的LiFePO4/C材料。在测试表征过程中,4个LiFePO4/C材料由于碳含量的差异表现出了不同的电化学性能,通过对测试数据的系统分析,得出主要结论如下:
(1)采用Li2CO3与LiOH·H2O为复合锂源合成LiFePO4/C材料,在高温下可以具有良好的熔融状态,所制得的LiFePO4/C材料具有优良的晶相结构与电化学性能;
(2)适当提高LiFePO4/C的碳包覆量能够改善材料的电导率,并有效地控制一次颗粒粒径从而提高锂离子扩散能力,改善动力学性能;
(3)LiFePO4/C材料碳含量为1%左右时,包覆量过低,导致材料的导电性差,体系内阻大,循环性能不佳,而当碳含量为3%与4%左右时,包覆量过高,导致材料的比表面积过大,体系副反应多,也使得循环性能恶化,长期存储后的界面稳定性变差。而包覆碳含量为1.95%的LFP-C-2样品,表面包覆状态良好,碳包覆层在LiFePO4颗粒表面均匀连续包覆,材料中不存在过剩的碳,表现出最优的电化学性能。
参考文献:
[1]Sun C W,Rajasekhara S,Goodenough J B,et al.J.Am.Chem.Soc.,2011,133(7):2132-2135
[2]Prosini P P,Zane D,Pasquali M.Electrochim.Acta,2001,46(23):3517-3523
[3]WANG Xiao-Jian(王小建),REN Jun-Xia(任俊霞),LI Yu-Zhan(李宇展),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21(2):249-252
[4]CHEN Yu(陈宇),WANG Zhong-Li(王忠丽),YU Chun-Yang(于春洋),et al.Acta Phys.-Chim.Sin.(物理化学学报),2008,24(8):1498-1502
[5]Wilcox J D,Doeff M M,Marcinek M,et al.J.Electrochem.Soc.,2007,154(5):A389-A395
[6]Recham N,Dupont L,Courty M,et al.Chem.Mater.,2009,21(6):1096-1107
[7]XIAO Zheng-Hui(肖正辉),LI Xue-Liang(李学良),CHEN Fei(陈飞),et al.Journal of the Chinese Ceramic Society(硅酸盐学报),2012,40(12):1823-1827
[8]ZHU Ling-Zhi(朱令之),WANG Chen-Xu(王晨旭),HAN En-Shan(韩恩山),et al.Chemical Industry and Engineering Progress(化工进展),2010,29(10):1927-1934
[9]Liu H,Li C,Zhang H P,et al.J.Power Sources,2006,159(1):717-720
[10]Murugan A V,Muraliganth T,Manthiram A.J.Phys.Chem.C,2008,112(112):46-54
[11]LU Jun-Biao(卢俊彪),TANG Zi-Long(唐子龙),ZHANG Zhong-Tai(张中太),et al.Journal of Inorganic Materials(无机材料学报),2005,20(3):666-670
[12]NI Jiang-Feng(倪江锋),ZHOU Heng-Hui(周恒辉),CHEN Ji-Tao(陈继涛),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21(4):472-476
[13]Wang L,He X M,Sun W T,et al.Nano Lett.,2012,12(11):5632-5636
[14]Huang Y H,Park K S,Goodenough J B.J.Electrochem.Soc.,2015,153(12):A2282-A2286
[15]Chueh W C,Gabaly F E,Sugar J D,et al.Nano Lett.,2013,13(3):866-872
[16]Liu H,Xie J Y,Wang K.J.Alloys Compd.,2008,459(1):521-525
[17]XIAO Zheng-Wei(肖政伟),HU Guo-Rong(胡国荣),DU Ke(杜柯),et al.Chinese Journal of Nonferrous Metals(中国有色金属学报),2007,17(12):2040-2045
[18]NI Jiang-Feng(倪江锋),ZHOU Heng-Hui(周恒辉),CHEN Ji-Tao(陈继涛),et al.Acta Phys.-Chim.Sin.(物理化学学报),2004,20(6):582-586
[19]Zhang X Y,Van H M,Singh D P,et al.Nano Lett.,2014,14(5):2279-2285
[20]Lu F,Zhou Y C,Liu J,et al.Electrochim.Acta,2011,56(24):8833-8838
[21]YU Feng(于锋),ZHANG Jing-Jie(张敬杰),YANG Yan-Feng(杨岩峰),et al.Chinese J.Inorg.Chem.(无机化学学报),2009,25(1):42-46
[22]Lin C J,Xu S C,Li Z,et al.J.Power Sources,2015,294:633-642
[23]Chang Y C,Peng C T,Hung I M.J.Mater.Sci.,2014,49(20):6907-6916
[24]Wang J J,Sun X L.Energy Environ.Sci.,2012,5(1):5163-5185
[25]Saravanan K,Reddy M V,Balaya P,et al.J.Mater.Chem.,2008,19(5):605-610
[26]Popovic J,Demircakan R,Tornow J,et al.Small,2011,7(8):1127-1135
