Ketamine enhances structural plasticity in human dopaminergic neurons: possible relevance for treatment-resistant depression
2018-05-05GinettaCollo,EmilioMerloPich
Depression refers to a series of mental health issues characterized by loss of interest and enjoyment in everyday life, low mood and selected emotional, cognitive, physical and behavioral symptoms. Depression is a common disorder, affecting 5–15% of the general population. When diagnosed as major depressive disorder (MDD), patients are currently treated with pharmacological agents such as serotonin or noradrenaline uptake inhibitors (SSRI or SNRI) or tricyclics. Patients that fail to respond to two or more antidepressant drugs - at an adequate dose for an adequate duration given sequentially – are considered affected by “treatment-resistant depression (TRD)”. TRD patients account for the 12–28% of the total MDD patients in charge to mental health services and are generally eligible to adjunctive pharmacotherapies with antipsychotics or lithium, behavioral therapies or electroconvulsive treatment (ECT). Unfortunately, several TRD patients display a persistent lack of response to the current adjunctive treatments, resulting in a relevant social burden and incurring in high direct medical costs to the health care system.
The biological underpinnings of TRD are partially unknown. One working hypothesis stipulates the occurrence of defective neuronal structural plasticity and adaptive molecular mechanisms in brain circuits that control emotion, cognition and motivation (Jernigan et al.,2011; Duman et al., 2016). Converging findings indicate that at least two neural circuits are involved: (1) the frontocortical-hippocampal system, characterized by pyramidal glutamatergic neurons projecting to other cortical and subcortical limbic structures (Duman et al.,2016); (2) the mesocorticolimbic dopaminergic (DA) system, whose DA neurons are located in the ventral mesencephalon and project to prefrontal cortex and limbic ventral striatum (Nestler and Carlezon,2006; Fawcett et al., 2016).
Defective functional and structural plasticity in the glutamatergic frontocortical and hippocampal circuits has been associated to high levels of circulating glucocorticoids and to reduced central levels of brain derived neurotrophic factor (BDNF) (Autry et al., 2011; Duman et al., 2016). Intriguingly, downregulation of intracellular molecular pathways involved in cell growth and survival,i.e., the mammalian target of rapamycin (mTOR) pathway, was described in postmortem prefrontal cortex of patients with mood disorders (Jernigan et al., 2011). Defective neuronal structural plasticity was also observed in frontocortical and hippocampal circuits of rodents after chronic stress, phenomenon reversed by repeated electroconvulsive therapy,chronic SSRI treatments or, as shown more recently, by acute infusion of ketamine (reviewed in Duman et al., 2016).
In patients with MDD/TRD, low sub-anesthetic doses of ketamine administered as single infusion for 45–60 minutes produce rapid antidepressant effects within hours (Zarate et al., 2006). The antidepressant properties of ketamine persist for several days, up to 2 weeks,beyond the duration of the acute psychotomimetic effects and the pharmacokinetic exposure. This profile requires neuroadaptation and,probably, a transient normalization of structural plasticity in the brain circuits involved in emotional, cognitive and motivational control(Duman et al., 2016).
To date the mechanism of the persistent antidepressant action of ketamine is only partially understood. Preclinical studies suggest that ketamine enhances glutamatergic neurotransmission in frontocortical and hippocampal pyramidal neurons. Ketamine, acting as non-competitive antagonist at the NR2B subunit of the heteromeric N-methyl-D-aspartate glutamate receptor (NMDAR), may attenuate the local gamma-aminobutyric acid (GABA) ergic interneuron activity resulting in glutamatergic pyramidal neurons disinhibition (Duman et al.,2016). The increase of glutamate neurotransmission strongly activates the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) localized post-synaptically, promoting, among other actions, the synthesis and release of BDNF (Autry et al., 2011). BDNF is known to drive protein synthesis and growth of dendrites and soma of pyramidal neurons in frontocortical and hippocampal circuits by binding to tropomyosin receptor kinase B (TrkB) receptors and activating the downstream mitogen-activated protein kinase kinase (MEK)-extracellular regulated protein kinases (ERK) and protein kinase B(Akt)-mTOR pathways, involved in cell survival and growth (Autry et al., 2011; Duman et al., 2016). This mechanistic interpretation involving the frontocortical-hippocampal system cannot properly explain some core symptoms of MDD/TRD such as anhedonia and lack of motivation, critically associated to a dysfunctional mesencephalic DA system (Nestler and Carlezon, 2006). In fact, drugs that enhance DA system neurotransmission,i.e., nomifensine and bupropion, are prescribed as antidepressants, while amphetamine is sometime used as a short term adjunctive treatment for TRD (Nestler and Carlezon,2006). Therefore it is reasonable to suggest that ketamine may exert its antidepressant effects acting also on the mesencephalic DA system.
Recent pharmacological data involving the mesencephalic system indicate that the D3 receptor (D3R)-preferential DA agonists pramipexole and ropinerole, originally developed as antiparkinsonian drugs,produce antidepressant effects (Breuer et al., 2009; Fawcett et al.,2016). D3Rs are expressed in ventral striatum and in mesencephalon,particularly in DA neurons as autoreceptors. In a series ofin vitroexperiments, using primary cultures of mouse mesencephalic DA neurons and iPSC derived DA neurons, we showed that D3R-preferential DA agonists and drugs that stimulate DA release (e.g., amphetamine)produce structural plasticity by increasing dendrite arborization and soma sizeviaD3R-mediated MEK-ERK and PI3K-Akt-mTOR pathway activation (Collo et al., 2008, 2018).
Recently, we investigated the influence of ketamine on DA system using a translationalin vitromodel by paralleling primary cultures of mouse mesencephalic DA neurons with cultures of human DA neurons differentiated from inducible pluripotent stem cells (iPSCs)of healthy volunteers, according to a standardized protocol (Cavalleri et al., 2017; Collo et al., 2018). Both humans and mouse cultures contained not only DA neurons but also GABAergic and glutamatergic neurons (Cavalleri et al., 2017). Cultures were exposed for 60 minutes to concentrations of ketamine similar to those producing antidepressant effects in humans and rodents. The effects on dendrite outgrowth and soma size were assessed 3 days after exposure, a time considered of relevance in modeling the sustained antidepressant effects of ketamine observed in the clinics.
The use of neurons differentiated from human iPSCs is a relatively recent acquisition for investigating the mechanism of action of neuroactive drugs (Avior et al., 2016; Cavalleri et al., 2017). Phenotypic explorations of neurons differentiated from human iPSCs have been increasingly recognized as a translational relevant approach for drug discovery in neurologic and psychiatric disorders (Avior et al., 2016;Collo et al., 2018). Using this approach, we also showed that ketamine produces structural plasticity in DA neurons by activating the PI3KAkt-mTORC1 pathwaysviaa BDNF/TrkB-dependent mechanism(Cavalleri et al., 2017). Similar effects were produced also by Ro25-6981, a selective NMDA NR2B subunit blocker. Visualization of increased phosphorylated p70S6 kinase in DA neurons indicated the recruitment of the mTOR pathway within minutes after ketamine exposure and a persistent increasevs. basal values for 60 min. Rapid phosphorylation of p70S6 kinase and long term structural plasticity were both blocked by the PI3K inhibitor LY294002 and the mTORC1 inhibitor rapamycin. Immuno-neutralization of BDNF, blockade of TrkB receptors and of MEK-ERK pathway likewise prevented ketamine-induced structural plasticity, confirming the involvement of BDNF/TrkB signaling in the activation of the mTOR also in DA neurons. Ketamine effects were abolished by the AMPA receptor antagonist NBQX as previously shown in mouse frontocortical and hippocampal neurons (Li et al., 2010; Autry et al., 2011). Intriguingly,we showed that ketamine-induced neuroplasticity on DA neurons required functionally-intact D3R, since its effects were abolished by selective D3R antagonist SB277011-A and were absent in primary cultures of DA neurons from D3R knockout mice (Cavalleri et al., 2017).
We also found that the ketamine metabolite 2R,6R-hydroxynorketamine (2R,6R-HNK), known for its longer pharmacokinetic half-life in humans, mimicked the structural plasticity effects of ketamine at sub-micromolar concentrations (Cavalleri et al., 2017). These data indicate that ketamine and its metabolite 2R,6R-HNK may elicit structural plasticity in DA neuronsviarecruitment of AMPAR and mTOR signaling in both mouse mesencephalic and human iPSC-derived DA neurons. This observation is of relevance for a better understanding of the antidepressant mechanism of action of ketaminein vivo.
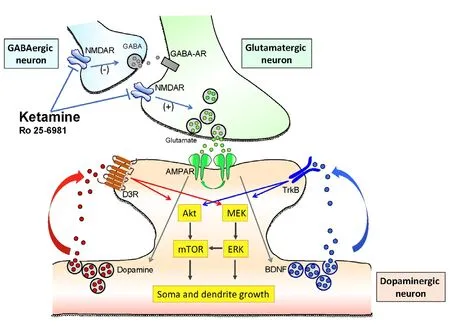
Figure 1 Cartoon representing the putative mechanism of action of ketamine and the molecular signalings involved in determining structural plasticity of dopaminergic neurons in vitro.
NMDAR: N-methyl-D-aspartate glutamate receptor; mTOR: mammalian target of rapamycin; ERK: extracellular regulated protein kinases;MEK: mitogen-activated protein kinase kinase; BDNF: brain derived neurotrophic factor; TrkB: tropomyosin receptor kinase B; GABA:gamma-aminobutyric acid.
The results hereby described could be integrated in one mechanistic working hypothesis represented in the cartoon ofFigure 1, in line with the working hypothesis proposed by Duman et al. (2016) for cortical neurons. According to this view, ketamine increases synaptic glutamate release by blocking NMDA receptors either on GABA interneurons, reducing their inhibitory input on the glutamatergic neurons, or at the glutamatergic synapsis, affecting the constitutive glutamatergic control of the excitatory synaptic drive. This may occur also for glutamatergic input on DA neurons. In addition, since NMDA receptors are also expressed on DA neurons, ketamine may have a potential direct effect. The “burst” increase of glutamate release induced by single infusion of ketamine activates the AMPA postsynaptic receptors on DA neurons (Figure 1), resulting in a three-fold effect:(1) the increase of translocation of novel AMPA receptors into the postsynaptic membrane, producing sensitization; (2) the increase of BDNF/TrkB signaling, possibly related to an increase of extracellular BDNF levels, and (3) the increase D3R signaling produced by an increased dopamine somato-dendritic release. These three mechanisms converge in the downstream activation of the MEK-ERK and AktmTOR pathways to drive the molecular machinery required for structural plasticity in dendrites and soma of DA neurons. It is tempting to use this working hypothesis to predict clinical therapeutic strategies in which ketamine and D3-preferring agonists could work synergistically to further improve the strength of induced neural plasticity, eventually prolonging the persistent antidepressant effects of ketamine (see also Collo et al., 2018).
Conclusion and summary:In this perspective article we summarized recent findings on the mechanism of action of ketamine focusing on mouse mesencephalic and human iPSCs-derived DA neurons. These results suggest that the prolonged antidepressant effects observed after a single infusion of ketamine in TRD patients can be related to a transient enhancement of structural plasticity induced by a glutamate “burst” occurring not only in frontal and hippocampal neurons but also in mesencephalic DA neurons. The known interconnectivity between these 2 circuits suggests a potential crosstalk. Since MDD/TRD has been associated to defective neural plasticity, the structural plasticity induced by these treatments could be interpreted as a potential remediation of an underlying neurobiological mechanism that sustains depressive symptoms. Intriguingly, the timing of thein vitroplasticity effects seems to be in line with the timing of the antidepressantin vivoeffects following a single ketamine infusion. While these conclusions await further confirmations, the present data support the use of iPSC derived DA neurons as a novel cellular translational model for drug discovery.
This work is funded by Ministry of Education, University and Research (MIUR)ex-60% research fund University of Brescia, Italy. Emilio Merlo Pich is employee of Takeda Pharmaceutical International AG. The Authors declare no conflict of interest.
Ginetta Collo*, #, Emilio Merlo Pich*, #Department of Molecular and Translational Medicine, Section of Pharmacology, University of Brescia, Brescia, Italy; Department of Biomedicine, University of Basel, Basel, Switzerland (Collo G)Neuroscience Therapeutic Area Unit, Takeda Pharmaceuticals International, Zurich, Switzerland; Division of Brain Science,Imperial College, London, UK (Merlo Pich E)
orcid:0000-0003-1888-5049 (Ginetta Collo)0000-0001-5478-235X (Emilio Merlo Pich)
#The authors contributed equally to this work.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-Share-Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer review reports:
Reviewer 1:Xiping Zhan, Howard University College of Medicine, USA.
Comments to authors:The review has focused on the work performed in primary dopaminergic neurons and human iPSC-induced dopamine neuron, and has thus added significant contribution to the role of dopaminergic involvement in depression.
Reviewer 2:Ulf Strauß, Charite Universitatsmedizin Berlin, Germany.
Comments to authors:This is a short recapitulation of the recent work on ketamine effects on human dopaminergic neurons. It presents a good view on recent progress in the field of depression research, where the development of truly novel medications is still somewhat hampered by our insufficient understanding of the pathophysiology. Here new rapid antidepressant agents such as ketamine have a huge impact, not only in terms of treatment but also by providing new insights in pathophysiological processes. This, in turn, will make the identification of new targets possible.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM(2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91-95.
Avior Y, Sagi I, Benvenisty N (2016) Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol 17:170-182.
Breuer ME, Groenink L, Oosting RS, Buerger E, Korte M, Ferger B, Olivier B (2009) Antidepressant effects of pramipexole, a dopamine D3/D2 receptor agonist, and 7-OHDPAT, a dopamine D3 receptor agonist, in olfactory bulbectomized rats. Eur J Pharmacol 616:134-140.
Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, Spano PF, Collo G (2017)Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry doi: 10.1038/mp.2017.241.
Collo G, Zanetti S, Missale C, Spano P (2008) Dopamine D3 receptor-preferring agonists increase dendrite arborization of mesencephalic dopaminergic neurons via extracellular signal-regulated kinase phosphorylation. Eur J Neurosci 28:1231-1240.
Collo G, Cavalleri L, Bono F, Mora C, Fedele S, Invernizzi RW, Gennarelli M, Piovani G,Kunath T, Millan MJ, Merlo Pich E, Spano P (2018) Ropinirole and pramipexole promote structural plasticity in human iPSC-derived dopaminergic neurons via BDNF and mTOR signaling. Neural Plast 2018:4196961.
Duman RS, Aghajanian GK, Sanacora G, Krystal JH (2016) Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med 22:238-249.
Fawcett J, Rush AJ, Vukelich J, Diaz SH, Dunklee L, Romo P, Yarns BC, Escalona R (2016)Clinical experience with high-dosage pramipexole in patients with treatment-resistant depressive episodes in unipolar and bipolar depression. Am J Psychiatry 173:107-111.
Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, Karolewicz B (2011) The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 35:1774-1779.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010)mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959-964.
Nestler EJ, Carlezon WA, Jr. (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59:1151-1159.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS,Manji HK (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856-864.
杂志排行

中国神经再生研究(英文版)的其它文章
- Acupuncture and neuroregeneration in ischemic stroke
- The adjustment of γ-aminobutyric acidA tonic subunits in Huntington’s disease: from transcription to translation to synaptic levels into the neostriatum
- Bridging the gap: axonal fusion drives rapid functional recovery of the nervous system
- Collagen for brain repair: therapeutic perspectives
- Stimulating effect of thyroid hormones in peripheral nerve regeneration: research history and future direction toward clinical therapy
- Harnessing migraines for neural regeneration