基于基因组学的前列腺癌临床精准诊疗研究进展
2018-05-04顾成元叶定伟
顾成元, 叶定伟
复旦大学附属肿瘤医院泌尿外科,复旦大学上海医学院肿瘤学系,上海 200032
前列腺癌是美国男性最常见的肿瘤之一,位居男性恶性肿瘤死亡率的第2位[1-2]。虽然前列腺癌的治疗已取得了长足的发展,已有多种用于治疗转移性前列腺癌的新药获美国食品与药品监督管理局(FDA)批准[3],但实现不同患者的精准化个体治疗仍存在极大的挑战。Young等[4]提出应在临床研究设计中联合生物学标志物检测来分层筛选患者。前列腺癌的分子分型可能有助于提高治疗效果,可以最大限度地减少无效治疗时间和不良反应等。高通量测序等新兴技术的应用有利于促进前列腺癌的分子分型,并为精准治疗的发展提供了依据。因此,本文就目前基于基因组学的前列腺癌分子分型相关研究及临床精准诊治研究进展作一综述,以切实提高前列腺癌的诊治水平。
1 Ets基因融合及其尿液检测试验对前列腺癌的诊断价值
研究[5]采用生物信息学方法检测基因芯片技术首次发现前列腺癌中存在的基因融合,其中最常见的染色体重排涉及雄激素调控基因TMPRSS2的5′非翻译区以及Ets转录因子(E26 transformation specific, Ets)家族成员Erg和Etv1。前列腺癌中这些基因融合的出现特异性近100%。进一步的研究[6]显示50%的前列腺特异性抗原(prostate specific antigen, PSA)筛查和15%~35%未筛查前列腺癌患者中存在Ets相关基因融合。最常见的融合基因发生于TMPRSS2和Erg基因之间,约占Ets基因融合的90%。回顾性研究[6]分析了基因融合与Gleason评分、病理分期和疾病特异性生存率的关系,但结果并不一致,可能由于研究人群和融合基因检测方法差异所致。Prensner等[7]研究也认为前列腺癌中融合基因的高度特异性具有潜在的诊断价值。
PSA检测广泛应用于前列腺癌的筛查,但其局限性包括假阳性及对惰性前列腺癌的过度诊断。为改善以PSA为基础的筛查,Tomlins等[8]开发了PSA联合尿液检测TMPRSS2-Erg融合基因和非编码RNA PCA3的方法来预测前列腺癌可能性。该方法可减少良性前列腺增生患者接受穿刺活检的比例,但需进一步的前瞻性研究来证明其有效性和特异性。
2 前列腺癌精准治疗相关生物学标志物的筛选与分析
分子标志物有助于区别惰性和侵袭性前列腺癌。目前已出现数个有前景的分子标志物,但受限于检测方法不统一、研究样本量小和验证研究缺乏等,尚未投入临床应用[7,9]。在前列腺癌前瞻性研究中需要延长随访时间观察复发风险也增加了研究的难度。新的高通量技术如蛋白质组学和基因组学使系统筛选分子标志物成为可能。近年来,核酸测序技术的迅速发展使肿瘤基因组大规模测序[10-11]、拷贝数变异、体细胞点突变、结构重排和基因表达改变的系统检测成为现实[12-13],为原发性和转移性前列腺癌基因组描绘了大致的轮廓。迄今为止,前列腺癌中最常见的分子生物学改变包括:雄激素受体(androgen receptor, AR)基因突变或扩增[14],Ets转录因子的基因重排[5],以及磷酸酶和张力蛋白同源物[PTEN/磷脂酰肌醇3-激酶(PI3K)]的丢失[15]。此外还包括发生频率较低(<5%)的异常相互排斥基因变化,如Akt激活[16]、PIK3CA和Ras突变以及BRAF突变或融合[17-18]。
2.1 高频基因组突变
2.1.1 Ets基因融合 由于前列腺癌中Ets基因融合的发生率约为50%,针对Ets基因融合的靶向治疗将使大量患者获益,但目前尚无直接针对此靶点的临床试验。与其他涉及转录因子的基因融合一样,相关靶向药物的开发步履维艰。但是如合成致死或阻断Ets转录因子的特定的蛋白间的相互作用等新技术在逐步完善之中[19]。同时,有研究[20-21]为研制以抑制聚ADP核糖聚合酶(PARP)或DNA-PK抑制的Ets转录因子相关复合物为基础的靶向药物提供了理论基础。此外,Barboro等[22]的研究结果显示PARP活性可调控下游的AR信号通路,为AR、PARP联合抑制提供了进一步的理论依据。数项关于转移性去势抵抗性前列腺癌 (castration-resistant prostate cancer, CRPC)的临床试验评估了PARP和DNA-PK抑制剂以及联合PARP和AR抑制的疗效,对Ets融合状态的理解将是研究的关键因素。
2.1.2 AR基因突变 目前前列腺癌基因组的数据表明超过50%的转移性CRPCs出现AR的扩增或突变。随着下一代针对雄激素信号通路靶向药物的开发,基因组测序策略将在确定潜在的耐药机制方面发挥作用。以雄激素合成和受体为靶点的联合治疗疗效仍未可知。
除了AR的突变和扩增,Dehm等[23]报道多种AR剪接异构体可导致雄激素信号通路的持续激活。这些异构体导致C-末端的配体结合域的损失,保存包含DNA结合及转录激活域的N-末端结构域和反式激活从而发挥作用。尽管有研究[24-25]已报道了多个亚型,二代测序技术发现了新的亚型,并且可能有助于理解其他潜在的耐药机制。转移性CRPC患者的AR剪接异构体发生频率尚未可知。因此,剪接体的相对意义以及它是否和其他耐药机制如突变或扩增等同时发生尚未明确,基因组改变如隐蔽的剪接位点缺失可能会影响剪接过程[26]。虽然目前没有以阻止AR剪接为靶点的治疗,但已有研究[27]尝试通过阻断包含的DNA结合结构域的N-末端结构域来抑制AR信号通路。可通过对转移性CRPC患者进行全面的基因组和转录组分析来研究AR剪接异构体和二代抗雄药物之间的相互作用。
2.1.3 PTEN和PI3K通路 PTEN是一种具有双特异磷酸酶活性的抑癌基因。PTEN蛋白可通过拮抗酪氨酸激酶等磷酸化酶的活性参与细胞调控,并负向调节PI3K-Akt-Mtor信号通路。因此,PTEN基因丢失或失活将导致PI3K信号通路的激活。多种肿瘤中广泛出现PTEN丢失,为PI3K通路抑制剂治疗肿瘤提供了理论依据[28]。在大鼠体内实验中已证实PTEN基因的杂合性缺失与其他癌基因可共同作用促进前列腺肿瘤的生长[29]。
在转移性CRPC患者中,点突变、缺失或重排等各种形式的PTEN缺失发生率至少在50%以上。这还未包括将其他功能性的PTEN失活机制如表观遗传学改变或翻译后调控。例如,Berger等[30]发现PTEN相互作用基因MAGI2的丢失或突变会导致功能性的PTEN失活。近期有研究[15]发现Erg融合与PTEN丢失导致雄激素信号通路激活之间的关系,在Erg融合的转基因小鼠模型中诱导PTEN杂合缺失将导致前列腺肿瘤的加速进展。该结果与基因组数据一致,表明PTEN缺失和Ets融合并不是相互排斥的事件。Carver等[15]表明抑制AR或PI3K通路将导致另一条通路的激活,该研究提示在临床试验中应同时抑制AR和PI3K信号通路。
2.2 低频和个体基因组突变
2.2.1 PIK3CA 最初在对结直肠肿瘤组织进行外显子测序后发现PIK3CA激活突变,随后在乳腺癌和卵巢癌等肿瘤中也发现了类似现象[31]。PIK3CA热点突变的功能研究[32]显示PI3K通路下游激活包括Akt磷酸化等,同时伴有细胞增殖等表型改变。
在前列腺癌中,PIK3CA基因突变发生的频率约为5%,拷贝数改变约为10%[33-34],而PTEN缺失可达到50%。在研发PI3K抑制剂时,主要问题为PIK3CA基因突变与PTEN缺失的亚型之间是否存在功能的差异。此外,PI3K抑制剂治疗PIK3CA突变前列腺癌后可能造成AR通路激活,因此对于这一亚型采取双重抑制可能使患者获益。
2.2.2 Akt Akt是一种丝氨酸/苏氨酸蛋白激酶,是PI3K通路的组成部分。对乳腺癌、结肠癌和卵巢癌组织中Akt1、Akt2和Akt3基因编码区进行测序后,发现Akt1存在激活突变[35]。该突变造成E17K氨基酸替换,在同源结构域中影响蛋白定位,在体内和体外试验中改变了转换效率。Boormans等[16]在188例前列腺癌中对Akt1基因进行测序,激活的E17K突变的比例约为1.4%。
2.2.3 Ras-Raf通路 BRAF基因编码Ras信号转导级联通路中的一种丝氨酸/苏氨酸蛋白激酶。有研究[36]假设Ras通路的下游可能存在致癌激酶突变,研究者在一系列肿瘤细胞系中对多个Ras通路基因进行外显子测序,在BRAF激酶结构域中发现一个频发突变V600E。该激活突变可导致Raf激酶持续活化。目前Raf和MEK抑制剂用于BRAF突变黑素瘤中的临床试验已取得积极的结果[2,37],还有一些其他抑制剂用于BRAF基因突变肿瘤治疗的研究在进行中。
对于前列腺癌,北美前列腺癌基因组的研究[38-39]发现典型的BRAF和KRAS突变占1%~2%。转录组测序发现除了点突变,涉及BRAF的基因融合发生率为1%~2%。体外实验中发现这些基因重排导致Raf激酶的表达和固有活性产生变化,对于Raf抑制剂是敏感的。转移性CRPC中KRAS基因融合发生率约为1%,体外敲除实验可抑制其发展[40]。因此,对于这种亚型的患者存在靶向治疗的可能,这也提示了低频驱动基因突变增加了治疗的复杂性,突变存在多样性。
3 基于基因组学的前列腺癌新发基因组改变
最新的前列腺癌测序研究发现许多潜在具有临床转化意义的新基因组改变。在原发性和转移性前列腺癌的两项研究[38-39]中,高通量测序发现了高频拷贝数变化和点突变。以下一些基因的改变值得注意:SPOP(13%),FOXA1(3%~4%),AURKA(神经内分泌前列腺癌中约占40%),MED12(4%~5%)和MAGI-2(1%),每个都有特定的生物学功能和潜在的临床意义。
SPOP基因编码产物为泛素连接酶复合物的亚基,在原发性前列腺癌的外显子测序研究[39]中发现其突变,突变频率约为15%,其他研究中为6%~15%。有趣的是,SPOP突变主要影响蛋白的底物结合区域。SPOP突变与Est重排互相排斥,这意味着这两种突变在前列腺癌发生中是单独的事件。体外实验中SPOP突变的转染对细胞增殖并无影响,但细胞侵袭增加。SPOP突变的前列腺癌可能代表一种新的分子亚型,有待进一步的生物学功能实验证实其是否具有潜在的治疗意义。
FOXA1基因编码产物是一种参与AR共调节的转录因子,其点突变影响DNA结合的叉头域[41]。体外实验中FOXA1突变促进肿瘤生长,负向调节雄激素信号通路。有研究[33]证实神经内分泌分化前列腺癌中MYC和AURKA发生共同扩增,并对AURKA抑制剂敏感。这为在AURKA扩增的前列腺癌中应用AURKA抑制剂治疗提供了理论基础,已成为通过测序技术将基础研究结果迅速转化为临床试验的优秀案例。
MED12编码一种中介体复合物和细胞周期蛋白依赖性激酶的亚基,在子宫肌瘤出现高频突变。在前列腺癌中该突变可能影响周期蛋白依赖性激酶功能。CHD1编码一种参与染色质重塑的解旋酶DNA结合蛋白,在前列腺癌组织芯片和二代测序研究[38-39]中发现其纯合缺失的发生频率为8%~10%。在正常前列腺上皮细胞中,下调CHD1将导致侵袭性增加但并不转化为癌。CHD1缺失与Ets重排是互相排斥的。
显然这些新发现的意义有待进一步的生物学实验来确定,这是通过基因组技术对前列腺癌进行分子分型的进步,有望在将来转化为临床应用。
4 基于基因组学的前列腺癌靶向治疗临床进展
前列腺癌基因组数据的临床转化工作已经开展(表1),目前进行的有针对雄激素信号通路、PI3K通路激活以及Ets基因家族重排的临床研究。2016年以Ets重排和表达为靶点的PARP抑制剂奥拉帕尼用于转移性CRPC获得FDA突破性疗法资格。在既往接受过一种基于紫杉烷的化疗及至少一种最新激素药物治疗的患者中,用于BRCA1/2或ATM基因突变转移性CRPC治疗。FDA授予突破性疗法资格的标准是初步临床证据显示该药物与现有治疗药物相比,可能对至少一种临床重要的终点有实质性改善。授予奥拉帕尼这一资格的决定基于Ⅱ期TOPARP-A试验的结果[42],从总体来看,研究涉及的50例患者中,既往均接受过多西他赛化疗。49例(98%)接受过阿比特龙和恩杂鲁胺治疗;29例(58%)接受过卡巴他赛治疗;16例对奥拉帕尼治疗有效(33%,95%CI 20~48);12例接受治疗超过6个月。经二代测序确认共有16例患者存在DNA修复基因相关缺陷(包括BRCA1/2、ATM和CHEK2基因),其中14例(88%)对奥拉帕尼治疗有反应。总体耐受性良好,贫血和乏力是最常见的不良事件,可见PARP抑制剂奥拉帕尼对于那些常规治疗方案无效且DNA修复基因存在缺陷的CRPC患者有很好的治疗效果[42]。随着第二代AR拮抗剂的研究逐渐深入,目前焦点集中在疾病进展时雄激素通路是否已完全阻断以及耐药的机制。此外,以Ets融合和雄激素受体N-端为靶点的临床前研究工作正在逐步开展。由于Ets融合与AR异常的发生频率较高,这些潜在的治疗方法将适用于大多数晚期前列腺癌患者。
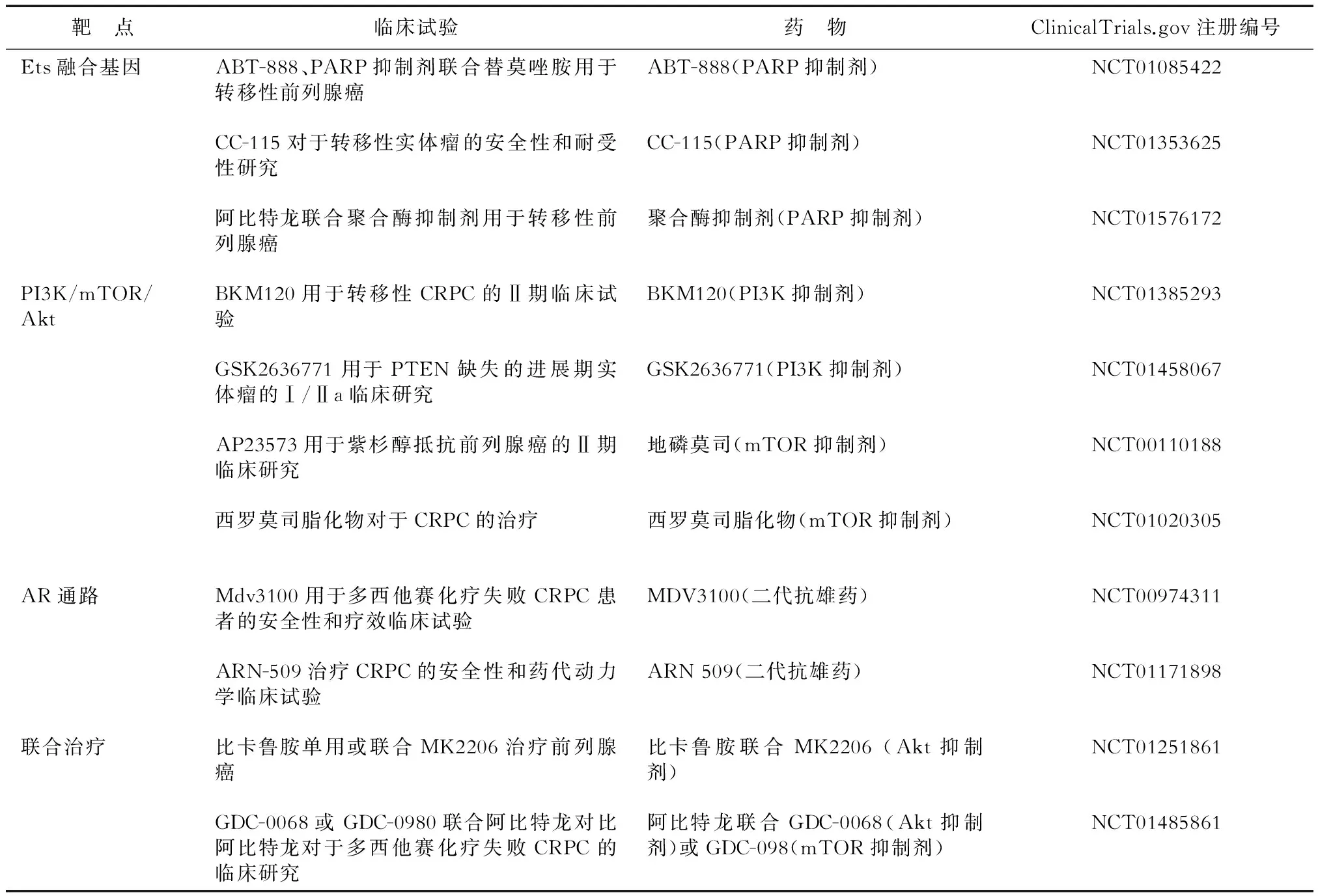
表1 前列腺癌靶向治疗临床试验一览表
基于联合阻断PI3K通路和雄激素信号通路的基础研究结果,已经开展相应的临床试验来验证这些假设。然而大部分此类试验未常规纳入综合分子分型,如PTEN和PI3K信号通路异常。治疗前和治疗后分别取活检进行系统分子分型将有助于发现和证实预测性生物标志物将使哪些患者获益。此外,基于活检的分子分型将提供组织信息为分析耐药机制并为下一步治疗或联合治疗提供依据。
对于具有低频和个体化变异的患者(1%~2%),地域和费用将对临床试验定期访视造成严重的困难。此外单中心研究的入组速度对制药企业来说可能过于缓慢。可能的解决方法是筛选时不局限于单个基因而是以信号通路为单位,并且入组任何病理类型的转移性前列腺癌。尽管这样的研究人群将出现异质性,但对于以探索为目的的研究是可以接受的。
为了促进这些罕见驱动突变患者的分子分型发展,研究人员已经开展了包括对转移性疾病组织活检进行二代测序等在内的肿瘤测序工作。临床肿瘤测序工作包括对肿瘤进行全基因组、外显子组或转录组测序,通过多学科合作对结果进行分析总结,提供具有临床操作性的结果反馈。近期有两个多中心研究[26]获得资助对转移性CRPC和黑素瘤患者进行临床肿瘤测序工作。这些工作将促进以分子分型为基础的精准医学概念逐步推广,寻找新的分子分型,有助于理解靶向治疗的耐药机制。
5 未来和挑战
虽然前列腺癌基因组数据逐步转化为临床应用,但仅采用高通量测序技术也有一定的局限性,包括组织活检、肿瘤微环境中,表观遗传学和异质性等也是需要考虑的因素。与其他上皮性肿瘤相比,相当一部分转移性前列腺癌患者仅有骨转移灶,这对于肿瘤组织活检是相当大的挑战。这对于组织采集和测序方法处理产生了限制。高达一半的骨组织活检取材量不足以用于测序分析。这些测序方法主要针对肿瘤细胞,可能忽略了肿瘤与宿主之间的相互作用,包括肿瘤微环境和免疫应答[43]。前列腺癌本身具有多病灶异质性的特点,关键问题是多个转移性病灶是否同样具有异质性。虽然这些病灶很可能都是驱动基因突变形成的优势克隆,但治疗方法和疗效是否将受到耐药性带来的异质性所影响目前仍不清楚。未来测序技术的进步,如单细胞测序等将有助于探索微环境的特征。而高通量测序的临床应用主要集中在DNA和RNA水平的变异,在表观遗传分析层面的分析尚未广泛开展。可用于预测预后的表观遗传变异和相应治疗非常有限。随着表观遗传学预测性生物标志物和相应治疗的发展和测序技术成本的逐渐下降,表观遗传分析也将加入到基础研究和临床应用中。
尽管存在局限性,未来十年中基因组分析仍将广泛应用于晚期前列腺癌的治疗。前列腺癌的分子特征促进了个性化肿瘤治疗临床试验的发展,前列腺癌基因组学的转化研究将促进临床试验设计的创新和尖端基础科学的临床应用。
[ 1 ] JEMAL A, SIEGEL R, WARD E, et al. Cancer statistics[J]. CA Cancer J Clin,2009, 59(4):225-249.
[ 2 ] FLAHERTY K T, ROBERT C, HERSEY P, et al. Improved survival with MEK inhibition in BRAF mutated melanoma[J]. N Engl J Med, 2012, 367(2):107-114.
[ 3 ] ATTARD G, DE BONO J S. Translating scientific advancement into clinical benefit for castrationresistant prostate cancer patients[J]. Clin Cancer Res,2011,17(12):3867-3875.
[ 4 ] YOUNG R C. Cancer clinical trials: a chronic but curable crisis[J]. N Engl J Med, 2010, 363(4):306-309.
[ 5 ] TOMLINS S A, RHODES D R, PERNER S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer[J]. Science,2005,310(5748):644-648.
[ 6 ] KUMAR-SINHA C, TOMLINS S A, CHINNAIYAN A M. Recurrent gene fusions in prostate cancer[J]. Nat Rev Cancer, 2008, 8(7):497-511.
[ 7 ] PRENSNER J R, RUBIN M A, WEI J T, et al. Beyond PSA: the next generation of prostate cancer biomarkers[J]. Sci Transl Med,2012,4(127):127rv3.
[ 8 ] TOMLINS S A, AUBIN S M, SIDDIQUI J, et al. Urine TMPRSS2: ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA[J]. Sci Transl Med,2011,3(94):94ra72.
[ 9 ] CHOUDHURY A D, EELES R, FREEDLAND S J, et al. The role of genetic markers in the management of prostate cancer[J]. Eur Urol, 2012, 62(4):577-587.
[10] PRENSNER J R, IYER M K, BALBIN O A, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression[J].Nat Biotechnol,2011,29(8):742-749.
[11] WANG Z, GERSTEIN M, SNYDER M. RNA-Seq: a revolutionary tool for transcriptomics[J]. Nat Rev Genet,2009,10(1):57-63.
[12] STRATTON M R, CAMPBELL P J, FUTREAL P A. The cancer genome[J]. Nature, 2009, 458(7239):719-724.
[13] MEYERSON M, GABRIEL S, GETZ G. Advances in understanding cancer genomes through secondgeneration sequencing[J]. Nat Rev Genet, 2010, 11(10):685-696.
[14] LINJA M J, SAVINAINEN K J, SARAMA O R, et al. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer[J]. Cancer Res, 2001, 61(9):3550-3555.
[15] CARVER B S, TRAN J, GOPALAN A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate[J]. Nat Genet,2009, 41(5):619-624.
[16] BOORMANS J L, KORSTEN H, ZIEL-VAN DER MADE A C, et al. E17K substitution in AKT1 in prostate cancer[J]. Br J Cancer, 2010, 102(10):1491-1494.
[17] CHO N Y, CHOI M, KIM B H, et al.BRAF and KRAS mutations in prostatic adenocarcinoma[J].Int J Cancer, 2006, 119(8):1858-1862.
[18] PALANISAMY N, ATEEQ B, KALYANA-SUNDARAM S, et al.Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma[J].Nat Med,2010, 16(7):793-798.
[19] KONSTANTINOPOULOS P A,PAPAVASSILIOU A G.Seeing the future of cancer-associated transcription factor drug targets[J]. JAMA, 2011, 305(22):2349-2350.
[20] BRENNER J C,ATEEQ B,LI Y,et al.Mechanistic rationale for inhibition of poly (ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer[J].Cancer Cell,2011, 19(5):664-678.
[21] GARNETT M J, EDELMAN E J, HEIDORN S J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells[J].Nature, 2012, 483(7391):570-575.
[22] BARBORO P, FERRARI N, CAPAIA M, et al. Expression of nuclear matrix proteins binding matrix attachment regions in prostate cancer. PARP-1: New player in tumor progression[J]. Int J Cancer, 2015, 137(7):1574-1586.
[23] DEHM S M, SCHMIDT L J, HEEMERS H V, et al.Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance[J]. Cancer Res, 2008, 68(13):5469-5477.
[24] DEHM S M, TINDALL D J.Alternatively spliced androgen receptor variants[J].Endocr Relat Cancer, 2011, 18(5):R183-R196.
[25] PERRY A S, WATSON R W, LAWLER M, et al.The epigenome as a therapeutic target in prostate cancer[J]. Nat Rev Urol, 2010, 7(12):668-680.
[26] ROYCHOWDHURY S, CHINNAIYAN A M.Advancing precision medicine for prostate cancer through genomics[J].J Clin Oncol, 2013, 31(15):1866-73.
[27] ANDERSEN R J, MAWJI N R, WANG J, et al.Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor[J].Cancer Cell, 2010, 17(6):535-546.
[28] CHALHOUB N, BAKER S J. PTEN and the PI3-kinase pathway in cancer[J]. Annu Rev Pathol, 2009, 4:127-150.
[29] DI CRISTOFANO A, DE ACETIS M, KOFF A, et al.Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse[J]. Nat Genet, 2001, 27(2):222-224.
[30] BERGER M F, LAWRENCE M S, DEMICHELIS F, et al.The genomic complexity of primary human prostate cancer[J].Nature, 2011, 470(7333):214-220.
[31] SAMUELS Y, WANG Z, BARDELLI A, et al. High frequency of mutations of the PIK3CA gene in human cancers[J]. Science, 2004, 304(5670):554.
[32] SAMUELS Y, DIAZ L A JR, SCHMIDT-KITTLER O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells[J].Cancer Cell,2015,7(6):561-573.
[33] BELTRAN H, RICKMAN D S, PARK K, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets[J]. Cancer Discov, 2011, 1(6):487-495.
[34] SUN X, HUANG J, HOMMA T, et al. Genetic alterations in the PI3K pathway in prostate cancer[J]. Anticancer Res, 2009, 29(5):1739-1743.
[35] CARPTEN J D, FABER A L, HORN C, et al.A transforming mutation in the pleckstrin homology domain of AKT1 in cancer[J]. Nature, 2007, 448(7152):439-444.
[36] DAVIES H, BIGNELL G R, COX C, et al.Mutations of the BRAF gene in human cancer[J]. Nature, 2002, 417(6892):949-954.
[37] CHAPMAN P B, HAUSCHILD A, ROBERT C, et al.Improved survival with vemurafenib in melanoma with BRAF V600E mutation[J].N Engl J Med,2011,364(26):2507-2516.
[38] GRASSO C S, WU Y M, ROBINSON D R, et al.The mutational landscape of lethal castration-resistant prostate cancer[J]. Nature, 2012, 487(7406):239-243.
[39] BARBIERI C E, BACA S C, LAWRENCE M S, et al.Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer[J]. Nat Genet, 2012, 44(6):685-689.
[40] WANG X S, SHANKAR S, DHANASEKARAN S M, et al.Characterization of KRAS rearrangements in metastatic prostate cancer[J]. Cancer Discov, 2011, 1(1):35-43.
[41] YU X, GUPTA A, WANG Y, et al.Foxa1 and Foxa2 interact with the androgen receptor to regulate prostate and epididymal genes differentially[J].Ann N Y Acad Sci,2005, 1061:77-93.
[42] MATEO J, CARREIRA S, SANDHU S, et al. DNA-repair defects and olaparib in metastatic prostate cancer[J]. N Engl J Med,2015,373(18):1697-1708.
[43] TAYLOR B S, SCHULTZ N, HIERONYMUS H, et al.Integrative genomic profiling of human prostate cancer[J].Cancer Cell, 2010,18(1):11-22.
