NPC1基因突变致小婴儿尼曼-匹克病1例
2018-04-28王丹丹程亚颖
王丹丹,程亚颖
(河北省人民医院 儿科,河北 石家庄 050051)
1 临床资料
患儿男性,1月12天。主诉:皮肤、黏膜黄染1个月余。查体:营养欠佳,全身皮肤黄染,无光泽,巩膜黄绿色,心肺查体无明显异常,腹澎隆,肝于肋下3.5 cm处扪及,质稍韧,边缘钝,脾于肋下约3.4 cm可触及,质中等,边缘钝,四肢肌张力尚可,吸吮、吞尚协调。辅助检查:肝功能TBIL:151.6 μmol/L、DBIL:118.5 μmol/L。血、尿及大便常规均无异常。肝胆动态显像结果示,肝外胆道通畅。TORCH抗体检测、肝炎相关病毒均阴性。腹部彩超示肝脾大。经保肝、消炎利胆治疗,皮肤黏膜黄染无减轻,肝脾进行性肿大。行外显子二代测序检测结果示,患儿NPC1基因c.2972-2973delAG和c.3382-3399del18 2个片段发生杂合变异,患儿父亲携带1个NPC1基因的杂合变异(c.2972-2973delAG位点),患儿母亲携带NPC1基因的杂合变异(c.3382-3399del18)(见图1~3),确诊尼曼-匹克病(Niemann-Pick disease,NPD)。家长放弃治疗,自动出院。后随访患儿出院后出现巩膜发蓝,腹泻、大便蓝色,于10个月龄死亡。
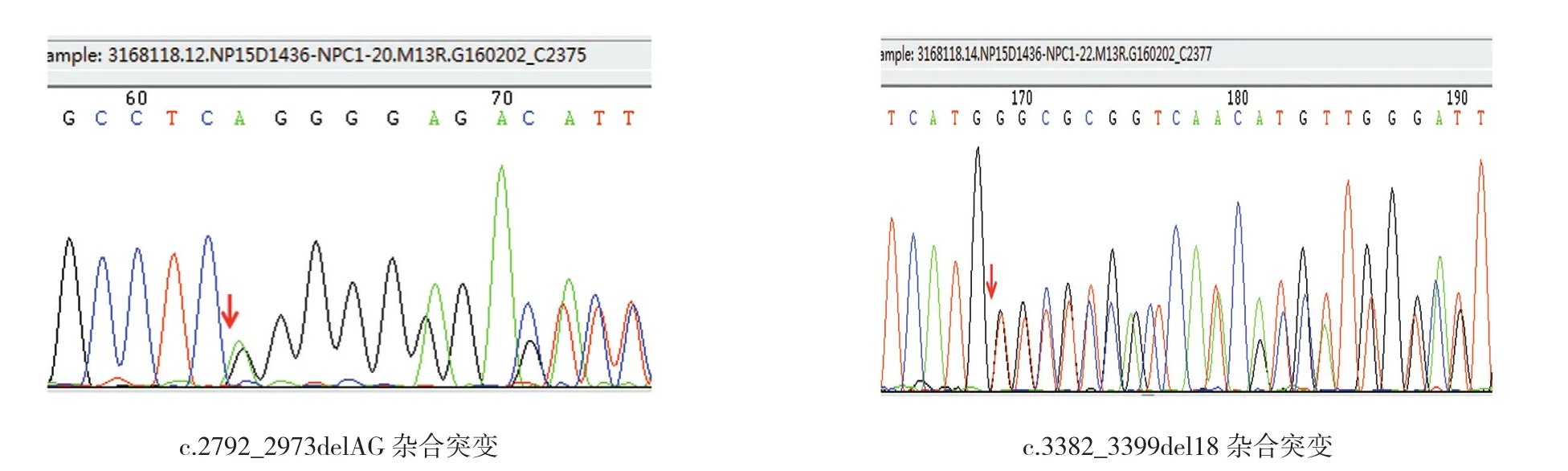
图1 受检者NPC1基因位点检测

图2 受检者之母NPC1基因位点检测

图3 受检者之父NPC1基因位点检测
2 讨论
2.1 概述
NPD系常染色体隐性遗传的先天性脂代谢紊乱性疾病,主要特点是神经鞘磷脂及胆固醇在单核-巨噬细胞系统和神经系统中大量沉积,形成特征性的泡沫细胞(尼曼-匹克细胞),导致内脏肿大、神经细胞死亡及白质髓鞘脱失等。临床特征包括肝脾肿大、肺功能不全和(或)中枢神经系统受损[1]。
2.2 临床表现
尼曼-匹克病临床表现多样,可分为5型(A~E型):A型(急性神经性),起病多在生后3~6个月,常表现为厌食呕吐、肝脾肿大、肌张力低及智力运动进行性减退等,严重可引起失聪、失明,或伴有贫血、恶病质,多因继发感染于学龄期前死亡。B型(慢性非神经型),多于婴幼儿或儿童期起病,病程进展相对缓慢,临床主要表现为肝脾肿大、脾功能亢进及肝功能和血脂异常等,无神经系统受损表现,智力运动发育正常。A型和B型均是位于第11号染色体(11p15)上SMPD1基因突变,使其编码的酸性鞘磷脂酶(ASM)活性低下或缺乏,导致鞘磷脂不能有效降解,堆积在肝脾、淋巴结、肾上腺皮质及骨髓中,形成富含脂质的尼曼-匹克细胞[2]。C型(慢性神经病型)自围产期至成人期均可发病,表现在内脏、神经、精神3大系统。内脏症状常见于4岁以下患儿,多表现为胆汁淤积性黄疸、进行性肝脾肿大[3]。神经症状常见于4岁以上的患儿[4],多表现为智能减退,构音、吞咽障碍,痫样发作,进行性共济失调及易跌跤等。精神症状多见于青少年及成人期,表现为偏执妄想、幻觉、认知障碍及攻击行为等。D型(Nova-Scotia型)具有与C型等位的缺陷基因,临床表现与C型相似,但病程进展缓慢,多数患儿发病于2~4岁。C型或D型是同时伴有第18号染色体(18q11)上NPC1和(或)第14号染色体NPC2基因突变,引起胆固醇转运障碍[5],使游离胆固醇在组织细胞内大量沉积。E型(成人非神经病型)极为少见,起病隐匿,表现为不同程度的肝脾肿大,智力正常,无神经系统受损表现。
2.3 诊断
NPD发病率低,易造成漏诊、误诊,其诊断需全面的了解病史,细致的体格检查,以及相关的实验室检查。临床疑似NPD的要点包括:①持续性黄疸、肝脾肿大;②精神运动发育迟缓或倒退;③其他系统相关表现,如:病因不明的失聪、失明,皮肤樱桃红斑,骨、关节疼痛或畸形等。确诊需要骨髓检查,肝脾、淋巴结活检及基因检测,有条件可行神经鞘磷脂酶活性的测定。该患儿新生儿期发病,以胆汁淤积、进行性肝脾肿大为主要表现,肝胆动态显像、TORCH抗体检测、肝炎病毒均无异常,为避免骨髓穿刺、淋巴结活检等有创操作,行基因检测。根据临床表现及基因检测结果,患儿确诊为尼曼-匹克病(C型)。
2.4 治疗
NPD尚无特异、疗效确切的方法,主要采用对症支持治疗。随着近年来国内外对该病的探索研究,有新的治疗方法逐渐在临床上应用。在欧盟、巴西等地区,美格鲁特(Miglustat)已作为治疗NPC型的首选药物,其可稳定并改善NPD-C患者的神经系统症状,延长预期寿命[6-7],且如能在神经系统症状出现之前开始使用,预后更好[8]。美国有报道提出用酶替代治疗NPD-B型患者,其动物模型为用重组人ASM替代治疗ASM基因敲除的小鼠,结果显示肝脾肺神经鞘磷脂含量降低,但神经系统症状无改善[9],适合用于非神经型尼曼-匹克病的治疗。
尼曼-匹克病作为一类发病罕见的代谢性疾病,临床表现多样,小婴儿症状不典型时,诊断尤其困难,要求临床医生应全面询问病史、详细体格检查,尽量完善相关检查,避免漏诊、误诊。本例患儿采用二代测序技术对相关疾病行早期筛查,检测出致病基因NPCI,明确诊断。二代测序技术可对疾病相关致病基因进行高通量测序,突变基因的检出率高,对于临床遗传异质性高的疾病,有利于早期筛查及诊断,尽早治疗。
参 考 文 献:
[1] SCHUCHMAN E H. Desnick RJ Types A and B Niemann-Pick disease[J]. Mol Genet Metab, 2017, 120(1/2): 27-33.
[2] STERN G. Niemann-Pick’s and Gaucher’s diseases[J].Parkinsonism Relat Disord, 2014, 20(Suppl 1): S143-S146.
[3] 胡亚美, 杨艳玲, 尼曼-匹克病[M] //胡亚美, 江载芳, 申昆玲,主编,诸福棠实用儿科学. 第8版. 北京: 人民卫生出版社,2015: 2304-2305.
[4] MENGEL E, PINEDA M, HENDRIKSZ C J, et al. Differences in Niemann-Pick disease Type C symptomatology observed in patients of different ages[J]. Mol Genet Metab, 2017, 120(3): 180-189.
[5] SUZUKI O, ABE M. Secondary sea-blue histiocytosis derived from Niemann-Pick disease[J] J Clin Exp Hematop, 2007, 47(1): 19-21.
[6] PATTERSON M C, VECCHIO D, PRADY H, et al. Miglustal for treatment of Niemann-Pick C disease: a randomised controlled study[J]. Lancet Neurol, 2007, 6(9): 765-772.
[7] 任守臣, 尼曼匹克病C型诊疗新进展[J]. 中国当代儿科杂志,2015, 17(5): 533-538.
[8] DI ROCCO M, DARDIS A, MADEO A, et al. Early miglustat therapy in infantile Niemann-Pick disease type C[J]. Pediatr Neurol, 2012, 47(1): 47-43.
[9] MIRANDA S R, HE X, SIMONARO C M, et al. Infusion of recombinant human acid sphingomyelinase into Niemann-Pick disease mice leads tu visceral,but not neurological, correction of the pathophysiology[J]. FASEB, 2000, 14(13): 1988-1995.
