水通道蛋白4和髓鞘少突胶质细胞糖蛋白抗体阳性患者临床分析
2018-04-26康新梅孙晓渤李静陈晨卢婷婷舒崖清杨晖王展航李小晶胡学强陆正齐邱伟彭立胜
康新梅 孙晓渤 李静 陈晨 卢婷婷 舒崖清 杨晖 王展航 李小晶胡学强 陆正齐 邱伟 彭立胜
水通道蛋白 4抗体 (aquporin 4 antibody,AQP4-lgG)是NMOSD的特异性生物标志[1-2]。2004年LENNON等[2]最先发现在视神经脊髓炎(neuromyelitis Optica,NMO)患者中存在部分患者AQP4-IgG阳性,提示NMO可能为一种自身免疫性疾病,随后的研究又将NMO扩大为NMOSD,其中包括 NMO、视神经炎(optic neuritis,ON)、横贯性脊髓炎(transverse myelitis,TM)等。髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)是一种仅表达于中枢神经系统髓鞘表层含量占0.05%的髓鞘蛋白成分,是免疫球蛋白超家族成员之一[3]。SATO等[4]报道,AQP4-lgG阴性的NMOSD中MOG-IgG阳性率为21.1%。该研究对NMOSD的发病机制提出了一个新的补充,即血清MOG-IgG阳性的NMOSD可能是区别于AQP4-IgG阳性的另一种独立的疾病,尽管它们可能存在共同的的临床表现。此外,MOG-IgG也在儿童ADEM及MS患者中出现[3,7]。有研究者提出,将MOG-IgG阳性的中枢神经系统免疫相关性疾病患者定义为 “MOG-IgG阳性脱髓鞘病”[5]。研究AQP4-IgG和MOG-IgG阳性的中枢神经系统自身免疫性疾病患者流行病学与临床特征,可为指导临床疾病诊疗及进一步探讨疾病发病机制提供依据。
1 对象与方法
1.1 研究对象 收集2013年9月至2016年12月由中山大学附属第三医院、中山大学中山眼科中心、广州妇女儿童医疗中心、广东三九脑科医院初步诊断为中枢神经系统免疫相关性疾病的2068例患者作为研究对象。最后诊断疾病类型包括MS、ADEM、NMOSD、抗NMDAR脑炎及未分类脱髓鞘疾病。诊断标准:MS患者均符合2010年McDonald MS诊断标准[6],ADEM的诊断遵从2012年IPMSSG提出了关于儿童ADEM的专家观点标准[6],NMOSD患者均符合2015年国际NMO诊断小组修订的NMOSD诊断标准[7],抗NMDAR脑炎患者均符合 Dalmau 标准(2016)[8]。
1.2 研究方法
1.2.1 临床资料收集 临床资料收集主要通过电子病历查询和电话随访两种途径,排除随访过程中失访和不同意信息收集的患者。临床资料主要包括患者流行病学、临床表现,实验室检查,MRI影像以及最终诊断等。目前临床多采用扩展功能障碍量表 (expand disability status scal,EDSS) 评估NMOSD患者的临床神经功能缺陷程度。
1.2.2 AQP4-IgG、MOG-IgG 检测(图 1) AQP4-IgG检测采用德国EUROIMMUN公司的检测试剂盒[4,9],实验方法为间接免疫荧光法。MOG-IgG检测采用转染细胞-间接免疫荧光法:将共表达人全长MOG和GFP的质粒转染至人胚肾293T(HEK293T)细胞中,24 h后,3%多聚甲醛固定15 min后,依次孵育患者血清及荧光二抗,利用间接免疫荧光方法检测患者血清中的MOG-IgG。血清样本的稀释比例为1:10。
1.3 统计学方法 采用 SPSS 16.0进行统计分析。符合正态分布的计量资料以(±s)表示,非正态分布的计量资料以中位数 (最小值,最大值)[M(range)],组间比较采用t检验、Mann-Whitney U检验,计数资料采用Chi-square检验。检验水准α=0.05。
2 结果
2.1 流行病学资料 2068例患者中,AQP4-IgG阳性681例,MOG-IgG阳性110例,双阳性4例,双阴性1273例。AQP4-IgG阳性患者中男65例,女616例,男女比例 1:9.5;平均发病年龄为(41.7±14.9)岁,其中 0~17 岁占 4.1%(28/681),18~40 岁占 41.7%(284/681),41~65 岁占 50.1%(341/681),66~82岁占 4.1%(28/681)。MOG-IgG 阳性患者中男53例,女 57例,男女比例 1:1.08;平均发病年龄为 (27.0±17.7)岁,其中 0~17岁占33.6%(37/110),18 ~40 岁 占 44.5% (49/110),41 ~65 岁 占20%(22/110),66-80 岁占 1.8%(2/110)。 AQP4-IgG阳性患者以青中年女性为主,MOG-IgG阳性患者以儿童及青年为主。两组间男女比例和发病年龄都存在统计学意义(表1)。
2.2 临床表现、诊断及预后比较 AQP4-IgG阳性患者首发症状中视神经炎占38.4%(86/223),脊髓炎 26.1%(59/223),极后区综合征 13.3%(30/223);MOG-IgG阳性患者中,视神经炎占53.5%(33/62),脊髓炎 37.9%(23/62), 极后区综合征7.7%(4/62)。AQP4-IgG阳性患者疾病病程为36个月,MOG-IgG阳性患者为42个月。96.3%(215/223)的AQP4-IgG阳性患者和 83.4%(52/62)的MOG-IgG阳性患者存在病情缓解复发的现象,前者年复发率为0.87%(0.22%~1.78%),后者为0.83%(0.31%~1.83%)。AQP4-IgG阳性患者临床预后EDSS评分3分,略高于MOG-IgG阳性患者临床预后EDSS评分2.5分(表2)。
表1 AQP4-IgG阳性与MOG-IgG阳性患者流行病学比较(±s)

表1 AQP4-IgG阳性与MOG-IgG阳性患者流行病学比较(±s)
1)与 MOG抗体组比较,经独立样本 t检验,P<0.05;2)与MOG抗体组比较,经Chi-square检验,P<0.05注:NMOSD:视神经脊髓炎谱系疾病,NMO:视神经脊髓炎,ON:视神经炎,TM:交叉性脊髓炎,MS:多发性硬化,ADEM:急性播散性脊髓炎,CIS:临床孤立综合征
基本特征最终诊断组别AQP4抗体MOG抗体n 681 110发病年龄(年)41.7±14.91)27.0±17.7男女比例1:9.481)1:1.08 NMO 28(42.14%)2)15(13.64%)NMOSD ON 13(19.68%)2)54(49.09%)TM 142(20.85%)2)3(2.73%)MS ADEM NMDAR脑炎 CIS 0(0)2)7(6.36%)0(0)2)6(5.45%)0(0)2)5(4.55%)118(17.33%)20(18.18%)
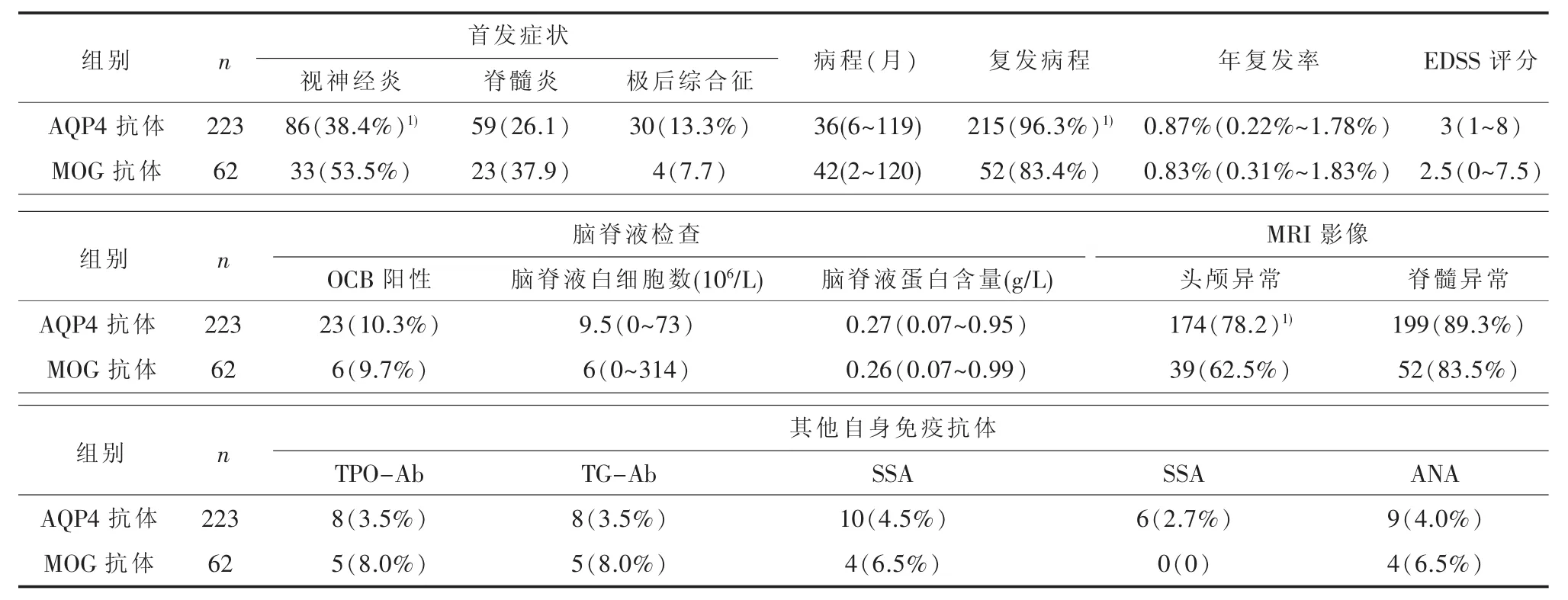
表2 AQP4-IgG阳性与MOG-IgG阳性患者临床特征比较[M(range))]
AQP4-IgG阳性患者最终诊断NMO占42.14%(287/681), 视神经炎 (ON)19.68%(134/681), 横贯 性 脊 髓 炎 (TM)20.85% (142/681),ODD17.33%(118/681);MOG-IgG 阳性患者最终诊 断 NMO 占 13.64%(15/110),ON 49.09% (54/110),TM 2.37% (3/110),MS 6.36% (7/110), ADEM 5.45%(6/110), 抗 NMDAR 脑炎 4.55%(5/110),ODD 18.18%(20/110)。 两组患者诊断分布在NMO、ON及TM的比例差异有统计学意义(P<0.0001)(表 1、图 2)。
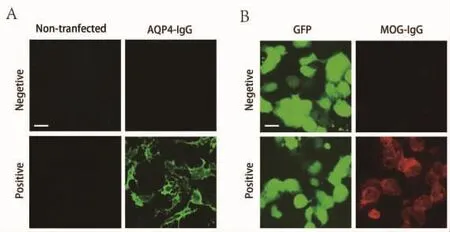
图1 AQP4-IgG(A)、MOG-IgG(B)荧光镜检结果图
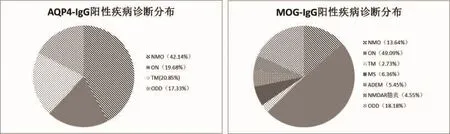
图2 AQP4-IgG阳性与MOG-IgG阳性患者疾病诊断分布比较
2.3 实验室检查及影像学资料比较(表2)对比两组患者的脑脊液检查发现,AQP4-IgG阳性患者脑脊液白细胞数 (×106/L) 为 9.5 (0~73), 略高于MOG-IgG阳性患者的脑脊液白细胞数 (106/L)为6(0~314);脑脊液蛋白含量(g/L)为 0.27(0.07~0.95),与MOG-IgG阳性患者脑脊液蛋白含量(g/L)为 0.26(0.07~0.99)无明显差异。共 23例(10.3%)AQP4-IgG阳性患者出现 OCB阳性,MOG-IgG阳性患者共有6例 (9.7%)为OCB阳性。两组之间脑脊液检查差异均无统计学意义。NMOSD可合并多种自身免疫性疾病,但也存在仅携带自身免疫抗体却不足以确诊相应疾病的现象。AQP4-IgG阳性患者中8例合并抗甲状腺过氧化物酶体(thyroid peroxisdase antibody, TPO)和抗甲状腺球蛋白抗体 (thyroglobulin antibody,TG)阳性,9 例合并抗核抗体(antinuclear antibody,ANA)阳性,10例合并SSA和6例SSB抗体阳性。MOG-lgG阳性患者中5例TPO和TG抗体阳性,4例合并ANA抗体阳性,4例合并SSA抗体阳性,5例合并NMDAR-lgG阳性。两组间差异无统计学意义。
MRI影像学资料显示,AQP4-IgG阳性患者中78.2%(174/223)存在头颅异常,较MOG-IgG阳性患者62.5%(39/62)常见;AQP4-IgG阳性患者89.3%(199/223)存在脊髓异常,与MOG-IgG阳性患者83.5%(52/62)差别不明显,但前者多以颈胸髓病灶多见,后者多累及胸腰髓;AQP4-IgG阳性患者99.8%(222/223)可累及长节段脊髓,稍高于MOG-IgG阳性患者96.3%(60/62)。两组患者MRI影像差别无统计学意义。
3 讨论
随着中枢神经系统自身免疫性疾病日渐受到关注,AQP4-IgG和MOG-IgG两种自身免疫性抗体的检测对中枢神经系统自身免疫性疾病的临床诊断具有重要意义。国外研究表明,AQP4-IgG阳性和MOG-IgG阳性患者可能存在流行病学及临床方面的差异,而国内目前尚无多中心大样本的分析研究。
国外学者SATO等[4]研究认为AQP4-IgG阳性患者中女性比例为87.7%(男/女=17/122),以女性患者为主;而MOG-IgG阳性患者中男性比例为62.5%(男/女=10/6),以男性患者多见;AQP4-IgG阳性患者平均发病年龄为32.5岁,略小于MOGIgG阳性患者平均发病年龄37岁。KITLEY等[10]在针对英国人群的研究中也有相似的发现。而在本项研究发现,AQP4-IgG阳性的NMOSD患者主要以女性多见,平均发病年龄为(41.7±14.9)岁,主要集中在青壮年时期,而MOG-IgG阳性患者男女比例基本持平,无明显分布差异,平均发病年龄为(27.0±17.7)岁,以儿童及青少年多见。
KITLEY等[10]研究发现,MOG-IgG阳性患者相比于AQP4-IgG阳性患者,更易发生双侧视神经炎 (bilateral optic neuritis,BON)或急性持续性ON;而SATO等[4]研究报道在BON和复发性ON患者中30.5%为AQP4-IgG阳性患者,27.8%为MOG-IgG阳性患者。本项研究显示,MOG-IgG阳性患者出现视神经炎的比例高于AQP4-IgG阳性患者,且多以双侧视神经炎多见;AQP4-IgG阳性患者出现脊髓炎少于MOG-IgG阳性患者;AQP4-IgG阳性患者较MOG-IgG阳性患者更易伴随极后区综合征。AQP4-IgG阳性患者诊断NMO和TM明显高于MOG-IgG阳性患者,ON在MOGIgG阳性患者的占比显著高于AQP4-IgG阳性患者。与AQP4-IgG阳性患者组不同,MOG-IgG阳性组患者最终诊断还包括MS、ADEM和抗NMDAR脑炎。其中 MS占 6.36%,ADEM占 5.45%,抗NMDAR脑炎占4.55%,ODD占18.18%。
AQP4-IgG阳性患者病程持续时间比MOGIgG阳性患者略短,但缓解复发性患者比MOGIgG阳性患者较多见。有研究报道,MOG-lgG阳性患者较AQP4-IgG阳性患者急性期及缓解期EDSS评分降低[11]。与本研究相似,本研究发现AQP4-IgG阳性患者疾病易反复,临床预后较差,而MOG-lgG阳性患者视力和运动恢复更好。
本研究发现,AQP4-IgG阳性患者脑脊液白细胞数和脑脊液蛋白含量都略高于MOG-IgG阳性患者,10.3%的AQP4-IgG阳性患者出现OCB阳性,略高于MOG-IgG阳性患者9.7%。NMOSD脊髓病灶多位于颈、胸髓内,长度大于3个椎体数[12]。有研究报道,MOG-IgG阳性患者颈椎比胸椎受损多见[13],而YAN等[14]则认为胸髓病灶略多于经髓病灶。与本研究相似,相比于AQP4-IgG阳性患者以颈胸髓受累常见,MOG-lgG阳性患者脊髓病灶多分布于胸髓,AQP4-IgG阳性患者更易出现头颅异常,且长节段脊髓受累的现象也稍常见。
国内学者报道在其收集的125例NMOSD患者中发现8%的患者为AQP4-IgG和MOG-IgG双阳性[14],国外采用CBA法检测的尚未发现两者双阳性现象。本项研究中发现4例AQP4-IgG和MOG-IgG共阳性患者。还有研究报道AQP4-IgG和MOG-IgG还可与其他自身免疫抗体存在共阳性。TITULAER等[15]报道存在MOG-IgG和NMDAR-IgG共阳性患者。PITTOCK等[16]报道AQP4-IgG 与抗核抗体(antinuclear antibody ,ANA)、干燥综合征A抗体(SSA)等存在共阳性。本项研究中,MOG-lgG阳性患者中5例合并抗甲状腺过氧化物酶体 (thyroid peroxisdase antibody, TPO-Ab)和抗甲状腺球蛋白抗体 (thyroglobulin antibody,TGAb)阳性,4 例合并抗核抗体(ANA)阳性,4 例合并SSA抗体阳性,5例合并NMDAR-lgG阳性。AQP4-IgG阳性患者中8例合并TPO-Ab和TGAb阳性,9例合并ANA抗体阳性,10例合并SSA和6例SSB抗体阳性。NMOSD可合并多种自身免疫性疾病,以干燥综合征(Sjögren's syndrome,SS)居多,SS常伴有神经系统受累,以周围神经系统受累较常见,且存在约20%患者伴有视神经炎[17]。NMOSD患者中合并自身免疫性甲状腺炎(autoimmune thyroid diseases,AITDs)常见于亚洲人群,其中以桥本甲状腺炎多见,常伴有TPO-Ab和TGAb升高,且抗体滴度与患者脊髓受损严重程度相关[18],还有研究发现NMO伴延髓损伤的患者常常也伴随出现AITDs,这类患者存在更严重的神经系统症状,且预后较差[19]。CHO等[20]认为NMO与多种自身免疫性疾病具有相同的遗传背景。NMOSD的病因及致病机制尚未清楚,但以上研究均提示伴随出现的其他自身免疫抗体是相应自身免疫病与NMOSD的重叠,而非NMOSD引起的疾病并发现象。希望本项研究结果能够为临床医生在诊治时提供一定的思路,从而制定更加精准的治疗方案。
综上所述,两组患者在流行病学、临床表现及诊断、预后上存在一定的差异,提示两组患者致病机制不同,在临床诊断及治疗上应加以区分。本项研究对两组患者临床资料的比较结果对临床疾病的诊断及鉴别诊断有一定指导意义。
[1]LENNON VA,KRYZER TJ,PITTOCK SJ,et al.IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel[J].J Exp Med,2005,202(4):473-477.
[2]LENNON VA,WINGERCHUK DM,KRYZER TJ,et al.A serum autoantibody marker of neuromyelitis optica:distinction from multiple sclerosis[J].Lancet,2004,364(9451):2106-2112.
[3]RAMANATHAN S,DALE R C,BRILOT F.Anti-MOG antibody:The history,clinical phenotype,and pathogenicity of a serum biomarker for demyelination[J].Autoimmunity Reviews,2016,15(4):307-324.
[4]SATO DK,CALLEGARO D,LANA-PEIXOTO MA,et al.Distinction between MOG antibody-positive and AQP4 antibodypositive NMO spectrum disorders[J].Neurology,2014,82(6):474-481.
[5]董会卿.MOG抗体介导的特发性炎性脱髓鞘疾病[J].中国神经免疫学和神经病学杂志,2017,24(3):88-91.
[6]Recommended Diagnostic Criteria for Multiple Sclerosis:Guidelines from the International Panel on the Diagnosis of Multiple Sclerosis[J].Ann Neurol,2001,50(1):121-127.
[7]REINDL M,DI PAULI F,ROSTASY K,et al.The spectrum of MOG autoantibody-associated demyelinating diseases[J].Nat Rev Neurol,2013,9(8):455-461.
[8]GRAUS F,TITULAER MJ,BALU R,et al.A clinical approach to diagnosis of autoimmune encephalitis[J].The Lancet Neurology,2016,15(4):391-404.
[9]董慧芳,张美妮,张静茹.髓鞘少突胶质细胞糖蛋白抗体阳性的视神经脊髓炎谱系疾病的研究进展 [J].中华临床医师杂志(电子版),2016(21):3286-3290.
[10]KITLEY J,WATERS P,WOODHALL M,et al.Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies:a comparative study[J].JAMA Neurol,2014,71(3):276-283.
[11]COBO-CALVO A,SEPULVEDA M,BERNARD-VALNET R,et al.Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis:Clinical and prognostic implications[J].Mult Scler,2016,22(3):312-319.
[12]WINGERCHUK DM,LENNON VA,LUCCHINETTI CF,et al.The spectrum of neuromyelitis optica[J].Lancet Neurol,2007,6(9):805-815.
[13]JARIUS S,RUPRECHT K,KLEITER I,et al.MOG-IgG in NMO and related disorders:a multicenter study of 50 patients.Part 2:Epidemiology,clinical presentation,radiological and laboratory features,treatment responses,and long-term outcome[J].J Neuroinflammation,2016,13(1):280.
[14]YAN Y,LI Y,FU Y,et al.Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD [J].Sci China Life Sci,2016,59(12):1270-1281.
[15]TITULAER M J,HOFTBERGER R,IIZUKA T,et al.Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis[J].Ann Neurol,2014,75(3):411-428.
[16]PITTOCK S J,LENNON V A,DE SEZE J,et al.Neuromyelitis optica and non organ-specific autoimmunity[J].Arch Neurol,2008,65(1):78-83.
[17]GONO T,KAWAGUCHI Y,KATSUMATA Y,et al.Clinical manifestations of neurological involvement in primary Sjogren's syndrome[J].Clin Rheumatol,2011,30(4):485-490.
[18]LONG Y,ZHENG Y,CHEN M,et al.Serum thyroid-stimulating hormone and anti-thyroglobulin antibody are independently associated with lesions in spinal cord in central nervous system demyelinating diseases[J].PLoS One,2014,9(8):e100672.
[19]WANG Y,ZHANG L,ZHANG B,et al.Comparative clinical characteristics of neuromyelitis optica spectrum disorders with and without medulla oblongata lesions[J].J Neurol,2014,261(5):954-962.
[20]CHO J H,GREGERSEN P K.Genomics and the multifactorial nature of human autoimmune disease[J].N Engl J Med,2011,365(17):1612-1623.
