“代谢记忆”与糖尿病肾病
2018-04-12熊晓燕综述白寿军审校
熊晓燕(综述) 白寿军(审校)
(复旦大学附属中山医院青浦分院肾内科 上海 201700)
糖尿病肾病(diabetic nephropathy,DN)是糖尿病患者最常见、最严重的慢性微血管并发症,其主要组织病理学特征是肾小球肥大、系膜增生、肾小管细胞外基质积聚所致的间质纤维化、基底膜增厚、足细胞缺失和足突融合[1]。目前已成为西方国家终末期肾脏病(end stage renal disease ,ESRD)的首位病因,也是我国 ESRD 的主要病因,仅次于肾小球肾炎。DN的发生发展机制错综复杂,多数研究认为,DN的发病多与遗传、糖化终末产物沉积、多元醇通路及蛋白质C激活、脂代谢异常、微循环障碍、氧化应激、炎症因子、激肽释放酶-激肽系统等有关。近年来的研究发现,表观遗传修饰异常[2]及足细胞自噬作用缺陷[3]在DN的发病及进展过程中也发挥着重要作用。约三分之一的糖尿病患者会发展为DN,使得DN的早期诊断和干预显得至关重要[1]。
“代谢记忆”概述“代谢记忆”的概念源自于包括糖尿病控制与并发症试验(Diabetes Control and Complications Trial,DCCT)及糖尿病干预与并发症流行病学(epidemiology of diabetes interventions and complications,EDIC)研究在内的一系列临床试验。DCCT试验发现,早期强化血糖控制的1型糖尿病患者较标准化血糖控制的患者肾脏、神经系统等并发症发生率及其严重程度明显降低。随后的EDIC 试验中,两组患者均使用强化治疗,尽管两组的HbA1c达到了相近水平,但之前在 DCCT中早期强化血糖控制的患者大血管及微血管并发症风险明显比未早期强化治疗的患者高,由此最早提出了“代谢记忆”这一概念[4]。其他以2型糖尿病患者为对象的试验亦发现,早期强化血糖控制在终止干预后仍能使患者长期获益,这一现象称之为“遗留效应”。“代谢记忆”现象在多种实验模型中亦得到了证实。El-Osta等[5]利用短时间的高糖培养基培养内皮细胞,发现即使在转为正常糖浓度培养后仍存在氧化应激持续激活和炎症因子高表达。Intine等[6]用0.3%的链脲佐菌素(streptozocin,STZ)溶液成功构建出了斑马鱼糖尿病代谢记忆模型。“代谢记忆”现象的提出使我们对包括DN在内的糖尿病并发症发生机制的认识有了突破性进展,大量的实验室及临床试验表明,“代谢记忆”现象的产生可能与晚期糖基化终末产物(advanced glycation endoproducts,AGEs)及氧化应激所诱发的瀑布式效应、过氧化亚硝酸盐的积累、表观遗传学修饰、慢性炎症及细胞凋亡等有关[7]。
“代谢记忆”现象与DN机体对早期高血糖的“记忆”效应对DN的发生发展起重要作用,关于其具体机制的研究在近年来的报道中屡见不鲜,然而确切的作用机制还不甚清楚。越来越多的研究证据表明靶细胞表观遗传学改变是产生高糖“记忆”、导致DN的重要原因[8-9]。表观遗传学作为连接“代谢记忆”与DN的“纽带”而备受关注。
表观遗传学修饰表观遗传学是遗传学中的一个前沿领域,主要包括DNA甲基化、组蛋白翻译后修饰、微RNA、染色质重塑等[10]。高血糖刺激后,靶细胞炎性介质表达失调、活性氧簇类过度蓄积、蛋白质硝基化增加、AGEs 生成增多、凋亡相关基因持续高表达,大量研究表明,这些因素失调与表观遗传修饰密切相关、相辅相成。表观遗传学标记稳定且可代际相传的特点,使得研究者们倾向于认为,表观遗传学变化可以为高糖“代谢记忆”提供分子机制方面的解释[11]。
DNA甲基化可阻断转录起始因子与启动子的结合而抑制基因转录。众所周知,足细胞为终末分化细胞,其结构和功能的完整性是维持正常肾功能的基础。研究发现许多对维持足细胞及裂孔隔膜完整性有重要作用的蛋白均受表观遗传学调控[12]。例如足细胞表达的转录因子(kruppel-like factor 4,KLF-4)在高糖等应激条件下可诱导体细胞重新编程转化为多能干细胞,从而发挥维持足细胞完整性作用,KLF-4因子与特定的反应元件结合时可导致nephrin启动子区CpG二核苷酸去甲基化及H3K9乙酰化、波形蛋白启动子区DNA甲基化,使nephrin蛋白表达增多,而波形蛋白表达减少。而研究发现在DN等一系列肾脏疾病状态下,KLF-4表达减少,这使得科学家们思考是否可利用转基因技术使KLF-4在足细胞中过表达以重建足细胞表型减少蛋白尿,Hayashi等[13]在多种动物模型中验证了这一设想。
组蛋白翻译后修饰包括乙酰化、磷酸化、甲基化、泛素化、ADP核糖化等[12],其中组蛋白甲基化及乙酰化在糖尿病“代谢记忆”和DN的进展中尤为重要。组蛋白转录后修饰可改变组蛋白与双链DNA的亲和力,从而改变染色质的疏松或致密状态,调节基因表达[14]。目前的研究认为,DN的发生与肾脏组蛋白H3K9及 H3K23的乙酰化、H3K4二甲基化、H3第10位丝氨酸磷酸化有关,这些修饰可使染色质解螺旋,促进基因表达[15]。DNA甲基转移酶(DNA methyltransferases,DNMTs)、组蛋白甲基转移酶(histone methyltransferases,HMTs)、组蛋白去甲基基酶(histone demethylases,HDMs)、组蛋白乙酰基转移酶(histone acetyl-transferases,HATs)、组蛋白去乙酰基酶(histone deacethylases,HDACs)等是药物开发的重要靶点。已有研究发现HDACs可使非组蛋白去乙酰化,HDAC4在人及啮齿类动物DN模型中均升高,敲除HDAC4同工型可减弱糖尿病大鼠足细胞损伤、蛋白尿及细胞外基质沉积,进一步的研究发现HDAC4敲除可重建被高糖削弱的足细胞自噬。研究者推测,其可能的机制是转录因子STAT1的去乙酰化抑制了足细胞自噬[16]。Gao等[17]发现高糖处理的肾小球系膜细胞组蛋白H2A泛素化增加而H2B泛素化减少,组蛋白泛素化水平的改变可激活TGF-β信号通路导致肾损伤,泛素蛋白酶体抑制剂MG132可通过抑制组蛋白异常泛素化抑制TGF-β通路激活。
miRNA是一种由大约22个核苷酸组成的非编码RNA,通过与特定的靶mRNAs 的3’-UTR结合来抑制基因表达,可参与维持细胞外基质动态平衡、保护足细胞功能。研究发现miRNA192、miRNA377、miRNA21和miRNA200家族等多种micro-RNA 与DN的发生有关。miRNA29c在db/db糖尿病小鼠肾小球系膜细胞中显著上调,而敲除miRNA29c则蛋白尿及系膜基质沉积明显减少[18]。最近的研究[19]显示,miRNA218可通过直接下调血红素加氧酶-1及促进p38-MAPK通路激活而加速高糖诱导的足细胞凋亡。miRNA125b、miRNA146a-5p及miRNA29a-3p的表达与NF-κB及p65等促炎基因的表达及持续性内皮功能损伤有关,是高糖“代谢记忆”的成因之一[20]。
以上这些机制并非完全独立,DNA甲基化与组蛋白修饰可相互作用,而这两者又可受miRNA的影响[21]。表观遗传学修饰既可使组织细胞或器官对环境刺激物迅速应答,又可在刺激物去除后使细胞产生“记忆”效应。
AGEsAGEs主要是由还原糖和蛋白质的氨基发生非酶促Maillard反应形成的稳定共价化物。AGEs的形成主要分3个步骤,其中最后一步为氧化、脱氢、环化反应。除血浆蛋白、脂蛋白等形成的AGEs外,大多数体内蛋白质形成的AGEs的半衰期都较长[22],可能是“代谢记忆”的成因之一。正常情况下,体内AGEs的产生与排出维持在一个较为平衡的状态,但当生成过多如糖尿病、衰老、摄入AGEs过多或肾脏排出障碍等情况下,便会产生一系列病理生理改变。一方面,葡萄糖等糖类物质直接与蛋白质和脂质交联,破坏蛋白甚至组织的结构和功能;另一方面,AGEs与其受体结合,激活一系列信号通路,增加炎症因子等的表达,最终改变组织细胞的功能,甚至产生组织破坏[23]。此外,有研究发现AGEs可通过氧化应激反应可引起胰岛β细胞合成及释放胰岛素功能障碍,诱导β细胞凋亡,还可抑制胰岛β细胞发生自噬作用从而促进胰岛β细胞凋亡[24]。AGEs可使肾小球系膜细胞发生非酶促糖化,导致肾小球高滤过。足细胞和肾小管细胞上表达的糖基化终末产物受体(receptor for advanced glycation end products,RAGEs)与AGEs作用后可通过促进活化氧类(reactive oxygen species,ROS)产生、减少内皮细胞NO产生等一系列反应导致肾小球硬化、肾小管间质纤维化[25]。IV型胶原及层粘连蛋白糖化可导致血管对蛋白质的渗透性升高,一些细胞外基质蛋白糖化可使其被基质金属蛋白酶的降解减少,导致基底膜增厚[26]。不论高血糖程度如何,AGE相关的细胞内线粒体呼吸链蛋白糖化可产生大量ROS,而ROS可通过非酶促糖化及糖氧化过程增加AGEs的生成,形成一个恶性循环放大AGE生成反应。这种瀑布式激活效应也是“代谢记忆”形成的重要原因[27]。
炎症糖尿病患者肾脏、血管、视网膜等靶器官中吞噬细胞浸润增加及炎症基因表达增强,表明慢性炎症是大多数血管并发症的共同特征。Chan等[28]用高糖处理与视网膜分离的周细胞糖6个月后发现IL-1、TNF、ICAM-1、VEGF、TGF-β等表达上调,再重新建立良好的血糖水平并不能完全降低先前这些炎症因子的上调及视网膜细胞的凋亡,说明周细胞对早期的高葡糖糖代谢产生了 “记忆”,炎症因子参与了“代谢记忆”现象的形成。已有研究[29]证实,高糖调控炎症基因表达增强主要与转录因子NF-κB通路激活有关,表观遗传学修饰参与了这一过程。NF-κB活化后诱导众多因子的转录,包括细胞生长因子、趋化因子、黏附分子和NF-κB抑制蛋白等,其中许多因子可增强循环中单核细胞的作用,使外周血循环中的单核细胞在血管内皮下聚集,是DN等糖尿病血微管病变发生的重要机制之一。在单核细胞内,高糖处理可招募组蛋白乙酰基转移酶(histone acetyl-transferases,HATs)如CBP/p300、p/CAF,进而促进炎症基因启动子区组蛋白H3赖氨酸乙酰化(H3Kac)、组蛋白H4赖氨酸乙酰化(H4Kac),导致染色质结构松散,促进基因转录[30]。在血管内皮细胞中,高糖主要通过招募组蛋白甲基转移酶SET7,促进炎症基因启动子区组蛋白H3的赖氨酸4单甲基化(H3K4me1),增加炎症基因及NF-κB活性亚基表达。
氧化应激及凋亡氧化系统与抗氧化系统失衡导致的氧化应激在DN的发生发展中起着关键性作用,高糖代谢产生的ROS是氧化应激导致组织损伤的中心环节[31]。线粒体是ROS产生的主要场所,高血糖时通过级联放大效应使线粒体氧化呼吸链产生过多的ROS,比如超氧阴离子(O2-)等,O2-是H2O2、过氧亚硝基及其他强活性氧类的前体,产生过多可造成线粒体损伤,线粒体是细胞内ATP生成的主要场所,其功能失调是DN等糖尿病并发症产生的重要原因。高糖刺激可促进线粒体被动力相关蛋白1(Drpl)和分裂蛋白(Fisl)切割为颗粒状,使得线粒体内呼吸链活性增强,线粒体膜通透性增加,ROS产生增加。另外,高糖刺激可使Rho相关的卷曲蛋白激酶1(Rho-associated,coiled-coil-containing protein kinase 1,ROCK1)激活,使Drp1-Ser600磷酸化而导致线粒体分裂,ROS产生增加[32]。ROS介导多元醇通路、己糖胺途径、RAGEs及其相关配体、蛋白激酶c等多个与DN相关的生物途径激活,这些代谢异常又可进一步产生ROS。ROS可造成DNA链断裂、碱基修饰、DNA-DNA交联和DNA-蛋白质交联等多种形式的DNA损伤,还可引起DNA甲基化状态改变,在表观遗传水平引起基因转录激活或抑制[33]。线粒体介导的细胞氧化损伤是肾小管上皮细胞凋亡的主要原因之一。线粒体ROS可激活p38丝裂原蛋白激酶(p38MAPK)及TGF-AP,促进肾小管上皮细胞转分化、间质细胞增生、细胞外基质聚集,最终导致肾纤维化。线粒体功能与内质网功能紧密相关,内质网应激亦是细胞损伤的重要原因。内质网是细胞内蛋白质合成折叠、钙离子储存、脂质合成的重要部位。正常情况下,内环境的稳定是内质网发挥正常功能的基本条件。在氧化应激导致蛋白质损伤时,可激活未折叠蛋白应答(unfolded protein response,UPR)以重建内质网稳态,而过度的UPR则可导致内质网应激(endoplasmic reticulum stress,ERS)使细胞功能失调并激活内质网跨膜蛋白肌醇酶1(inositol-requiring enzyme 1,IRE1)等促凋亡因子[34]。抗氧化应激药物、Drp1及ROCK1基因敲除可抑制线粒体分裂,减少ROS产生,是延缓“代谢记忆”和DN进展的潜在靶点。此外,蛋白质C是内皮细胞表面的一种酶原,当其被凝血调节酶/凝血蛋白激活后可抑制血液凝固、调节细胞内信号,具有细胞保护功能。Isermann等[35]发现,依赖凝血调节蛋白激活的蛋白质C被破坏后,糖尿病小鼠足细胞凋亡增加、氧化应激加剧、蛋白尿增多。那么蛋白质C抗氧化应激、行使细胞保护功能的机制何在?这一问题引发了研究者们的进一步探索。Bock等[36]发现暴露于高糖浓度的足细胞氧化还原酶 p66Shc呈蛋白质C依赖型表达。p66Shc可从细胞色素c接受电子参与线粒体氧化应激。他们进一步研究发现p66Shc高表达与其启动子区CpG二核苷酸过度甲基化及组蛋白H3高度乙酰化有关。这些研究不仅为氧化应激参与糖尿病足细胞损伤提供了有力证据,同时也证明氧化应激与表观遗传调控密切相关,为抗氧化应激治疗DN提供了实验依据。
结语“代谢记忆”与DN并不是简单的因果关系或平行关系,产生“代谢记忆”现象的内在机制是包括DN在内的一系列糖尿病并发症的基础。这些机制主要包括表观遗传学修饰、糖化终末产物、氧化应激、炎症等,各因素间相互作用造成糖尿病并发症发生发展(图1)。对于DN等糖尿病相关并发症除了早期强化血糖控制,减弱高糖“代谢记忆”效应外,还应针对以上机制选择合理有效的治疗方案。由于表观遗传学可在基因转录水平对各种疾病相关因子进行调控,因而成为近年来的研究热点。与经典遗传学不同的是,表观遗传信息主要决定组织细胞的功能状态,其调控作用的可逆性为表观遗传药物应用于临床提供了理论依据,目前已有针对肿瘤DNA异常甲基化及组蛋白异常修饰的表观遗传学药物批准上市。表观遗传及“代谢记忆”机制研究的不断深入将为DN的早期诊断及延缓肾功能恶化打下坚实基础。
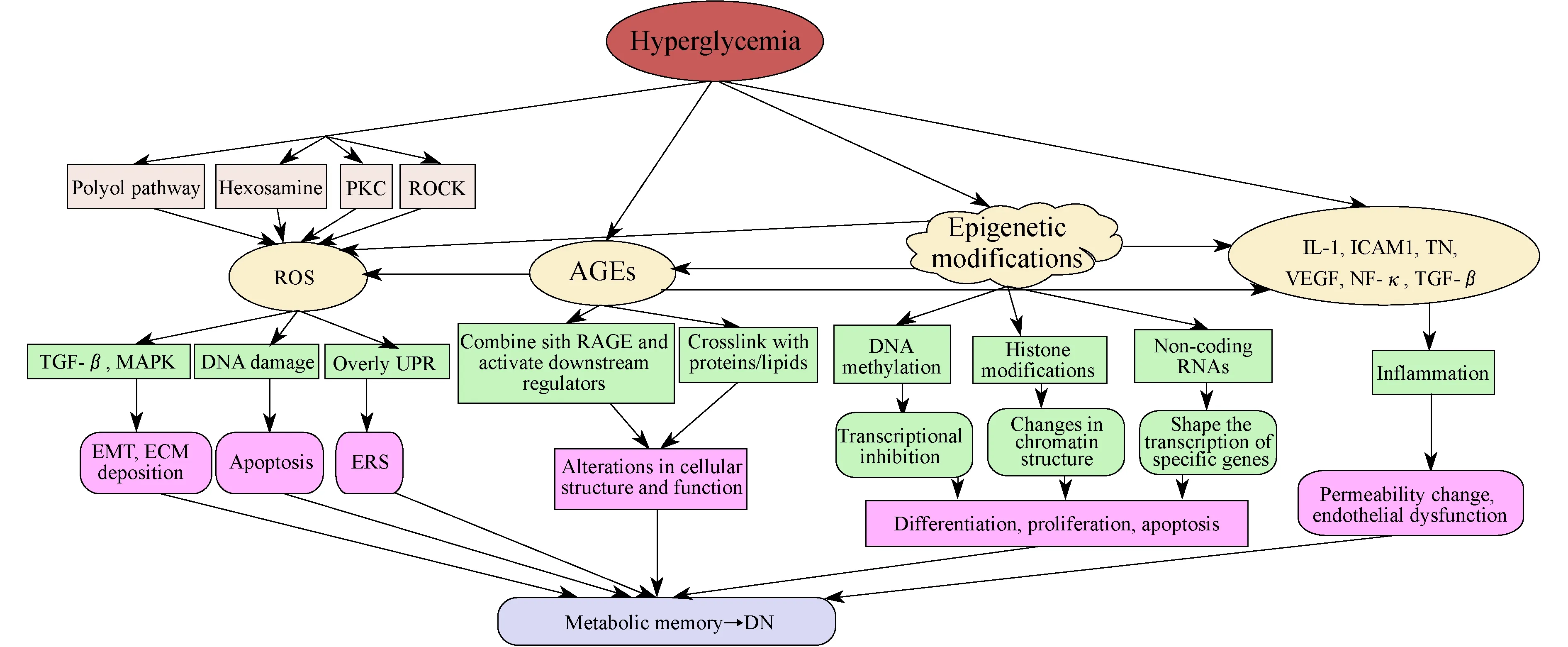
图1 高糖“记忆”与糖尿病发生发展的关系Fig 1 The relationship between hyperglycemic memory and diabetic nephropathy
[1]POURGHASEM M,SHAFI H,BABAZADEH Z.Histological changes of kidney in diabetic nephropathy[J].CaspianJInternMed,2015,6(3):120-127.
[2]KEATING ST,EL-OSTA A.Glycemic memories and the epigenetic component of diabetic nephropathy[J].CurrDiabRep,2013,13(4):574-581.
[3]TAGAWA A,YASUDA M,KUME S,etal.Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy[J].Diabetes,2016,65(3):755-767.
[4]CHEN Z,MIAO F,PATERSON AD,etal.Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort[J].ProcNatlAcadSciUSA,2016,113(21):E3002-E3011.
[5]EL-OSTA A,BRASACCHIO D,YAO D,etal.Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia[J].JExpMed,2008,205(10):2409-2417.
[6]INTINE RV,OLSEN AS,SARRAS MP JR.A zebrafish model of diabetes mellitus and metabolic memory[J].JVisExp,2013(72):e50232.
[7]BEREZIN A.Metabolic memory phenomenon in diabetes mellitus:Achieving and perspectives[J].DiabetesMetabSyndr,2016,10(2 Suppl 1):176-183.
[8]KATO M,NATARAJAN R.Diabetic nephropathy--emerging epigenetic mechanisms[J].NatRevNephrol,2014,10(9):517-530.
[9]TONNA S,EL-OSTA A,MARK EC,etal.Metabolic memory and diabetic nephropathy:potential role for epigenetic mechanisms[J].NatRevNephrol,2010,6(6):332-341.
[10]THOMAS MC.Epigenetic mechanisms in diabetic kidney disease[J].CurrDiabRep,2016,16(3):31.
[11]CENCIONI C,SPALLOTTA F,GRECO S,etal.Epigenetic mechanisms of hyperglycemic memory[J].IntJBiochemCellBiol,2014,51(4):155-158.
[12]MAJUMDER S,ADVANI A.The epigenetic regulation of podocyte function in diabetes[J].JDiabetesComplications,2015,29(8):1337-1344.
[13]HAYASHI K,SASAMURA H,NAKAMURA M,etal.KLF4-dependent epigenetic remodeling modulates podocyte phentypes and attenuates proteinuria[J].JClinInvest,2014,124(6):2523-2537.
[14]GILBERT RE,HUANG Q,THAI K,etal.Histone deacetylase inhibition attenuates diabetes-associated kidney growth:potential role for epigenetic modification of the epidermal growth factor receptor[J].KidneyInt,2011,79(12):1312-1321.
[15]NOH H,OH EY,SEO JY,etal.Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury[J].AmJPhysiolRenalPhysiol,2009,297(3):729-739.
[16]WEI Q,DONG Z.HDAC4 blocks autophagy to trigger podocyte injury:non-epigenetic action in diabetic nephropathy[J].KidneyInt,2014,86(4):666-668.
[17]GAO C,CHEN G,LIU L,etal.Impact of high glucose and proteasome inhibitor MG132 on histone H2A and H2B ubiquitination in rat glomerular mesangial cells[J].JDiabetesRes,2013,2013:589474.
[18]LONG J,WANG Y,WANG W,etal.MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1,and itsinvivoknockdown prevents progression of diabetic nephropathy[J].JBiolChem,2011,286(13):11837-11848.
[19]YANG H,WANG Q,LI S.MicroRNA-218 promotes high glucose-induced apoptosis in podocytes by targeting heme oxygenase-1[J].BiochemBiophysResCommun,2016,471(4):582-588.
[20]ZHONG X,LIAO Y,CHEN L,etal.The microRNAs in the pathogenesis of metabolic memory[J].Endocrinology,2015,156(9):3157-3168.
[21]LIN C,LEE PH,SHU YC,etal.MicroRNA-29a promotion of nephrin acetylation ameliorates hyperglycemia-induced podocyte dysfunction[J].JAmSocNephrol,2014,25(8):1698-1709.
[22]LAPOLLA A,BRIOSCHI M,BANFI C,etal.Nonenzymatically glycated lipoprotein ApoA-I in plasma of diabetic and nephropathic patients[J].AnnNYAcadSci,2008,1126:295-299.
[23]NISHIKAWA T,ARAKI E.Involvement of advanced glycation end-products in ‘hyperglycemic memory’[J].JDiabetesInvestig,2016,7(3):297-299.
[24] ZHU Y,SHU T,LIN Y,etal.Inhibition of the receptor for advanced glycation end products (RAGE) protects pancreatic beta-cells[J].BiochemBiophysResCommun,2011,404(1):159-165.
[25]BUSCH M,FRANKE S,RUSTER C,etal.Advanced glycationend-products and the kidney[J].EurJClinInvest,2010,40(8):742-755.
[26]SERBAN AI,STANCA L,GEICU OI,etal.RAGE and TGF-beta1 cross-talk regulate extracellular matrix turnover and cytokine synthesis in ages exposed fibroblast cells[J].PLoSOne,2016,11(3):e0152376.
[27]CHILELLI NC,BURLINA S,LAPOLLA A.AGEs,rather than hyperglycemia,are responsible for microvascular complications in diabetes:a “glycoxidation-centric” point of view[J].NutrMetabCardiovascDis,2013,23(10):913-919.
[28]CHAN PS,KANWAR M,KOWLURU RA.Resistance of retinal inflammatory mediators to suppress after reinstitution of good glycemic control:novel mechanism for metabolic memory[J].JDiabetesComplications,2010,24(1):55-63.
[29]REDDY MA,NATARAJAN R.Epigenetic mechanisms in diabetic vascular complications[J].CardiovascRes,2011,90(3):421-429.
[30]REDDY MA,ZHANG E,NATARAJAN R.Epigenetic mechanisms in diabetic complications and metabolic memory[J].Diabetologia,2015,58(3):443-455.
[31]KANWAR YS,WADA J,SUN L,etal.Diabetic nephropathy:mechanisms of renal disease progression[J].ExpBiolMed(Maywood),2008,233(1):4-11.
[32]WANG W,WANG Y,LONG J,etal.Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells[J].CellMetab,2012,15(2):186-200.
[33]FENG B,RUIZ MA,CHAKRABARTI S.Oxidative-stress-induced epigenetic changes in chronic diabetic complications[J].CanJPhysiolPharmacol,2013,91(3):213-220.
[34]LINDBLOM R,HIGGINS G,COUGHLAN M,etal.Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy[J].RevDiabetStud,2015,12(1-2):134-156.
[35]ISERMANN B,VINNIKOV IA,MADHUSUDHAN T,etal.Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis[J].NatMed,2007,13(11):1349-1358.
[36]BOCK F,SHAHZADA K,WANG H,etal.Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc[J].ProcNatlAcadSciUSA,2013,110(2):648-653.
