昔康衍生物的设计、合成和体外抗肿瘤活性研究
2018-04-10李家明张恩立
王 杰 李家明 张恩立 张 晖
(1.蚌埠医学院公共基础学院 安徽蚌埠 233030;2.安徽中医药大学药学院 安徽合肥 230031)
肿瘤是目前全球所面临的最棘手的公共卫生问题之一,它对人类健康具有较严重的危害。近年来,随着人口老龄化的加快和环境污染的加重,我国肿瘤的发病率呈逐年上升趋势[1,2]。而传统细胞毒类抗肿瘤药物在抑制肿瘤细胞的同时对正常细胞也具有较强的抑制作用,因此开发选择性高、疗效佳的靶向抗肿瘤药成为了国际研究的热点[3,4]。
美洛昔康、吡罗昔康(图1)是临床中常用的一类具有苯并噻嗪环结构的非甾体抗炎药,具有镇痛、消炎、抗血栓、抑制肿瘤细胞增殖等药理活性[5,6]。辛兵[7]等通过体内体外实验研究表明,美洛昔康对卵巢癌细胞株(OVCAR-3、HTOA) 的抑制作用明显优于顺铂,且可促进细胞的凋亡。Thimmaraju[8]等研究表明,美洛昔康/泊洛沙姆407凝胶制剂对HL-60细胞株具有显著的抑制作用,GI50<10μg/ml。本课题组对美洛昔康等苯并噻嗪类化合物的结构进行剖析发现,该类化合物分子中的羟基和酰胺羰基可通过氢键形成一个稳定六员环,该构型和EGFR受体抑制剂吉非替尼的母核结构相似(图2),相关工作见文献[9]。对本课题组前期研究总结时发现,含有-CF3基团的苯并噻嗪衍生物对肿瘤细胞A549的抑制活性优于阳性对照组药gefitinib。
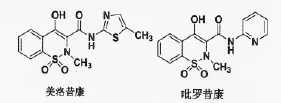
图1 美洛昔康、吡罗昔康的结构
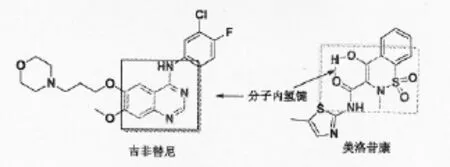
图2 吉非替尼和昔康类化合物的结构
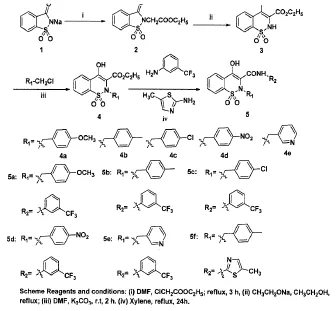
本文依据生物电子等排原理和骨架跃迁原理,设计并合成了5个含-CF3基团和1个含噻唑基团的昔康衍生物,路线如Scheme,所合成的6个化合物的MTT实验结果显示,部分化合物对A549、A431具有较好的抑制活性。
一、实验部分
(一)仪器与试剂。LCQADVANTAGE MAX液质连用质谱仪 (FINNIGA公司);Bruker 300 MHz超导核磁共振仪(DMSO-d6为溶剂,TMS为内标);FA2004B电子天平(上海平轩科学仪器有限公司);CO2培养箱(3111,美国SHELLAB公司);倒置光学显微镜(TS100-FDH1,NILON 公司);酶标仪(SpectramaxM2e,Molecular Devices公司)。
3-氯甲基吡啶盐酸盐、氯乙酸乙酯、4-甲基氯化苄、糖精钠、3-三氟甲基苯胺、2-氨基-5-甲基噻唑购自上海毕得医药科技有限公司;4-甲氧基氯化苄、4-氯苄氯、乙醇钠由本实验室自制;吉非替尼、美洛昔康由合肥医工医药有限公司提供;乙醇、二甲苯、DMF等常规试剂均购自合肥宝添科贸有限公司。
(二)化合物的合成。
1.3-氧代-1,2-苯并异噻唑-2-乙酸乙酯1,1-二氧化物(2)的合成。向一250mL三口烧瓶中加入30g糖精钠(0.15mol)、18g氯乙酸乙酯(0.15mol)和 80mLDMF,磁力搅拌下,缓慢升温至120℃反应3h,经TLC[V(石油醚):V(乙酸乙酯)=1:1为展开剂]检测反应结束后,水浴冷却至25℃,搅拌下一次性加入250mL水,立即析出白色固体,于冰浴下静止1h,抽滤,滤饼用冷水(50mL×3)洗涤,用80%乙醇重结晶,干燥,得白色固体化合物(35.3g),收率 87.5%,m.p.103~105℃。(文献[10]m.p.106~108℃)。
2.4-羟基-2H-1,2-苯并噻嗪-3-羧酸乙酯1,1-二氧化物(3)的合成。向一装有机械搅拌和温度计的250mL三口烧瓶中加入60mL无水乙醇和2.6g被切成小块状的金属钠(0.11mol),搅拌至钠块完全溶解后,将温度升高至90℃,一次性快速加入10g自制干燥固体化合物2,搅拌0.5h后,反应温度调至60℃,继续搅拌2h,TLC[V(石油醚):V(乙酸乙酯)=2:1为展开剂]检测反应结束后,将温度降至5~10℃,机械搅拌下,一次性加入碎冰(72g)和浓盐酸(28mL)混合液,析出白色固体,撤去搅拌将三口瓶放置冰盐浴下1h,抽滤,滤饼用冷水洗涤(30mL×3),干燥得白色化合物 3(5.5g),收率为 55.0%,m.p.167~169℃(文献[11]:m.p.169~170℃)。
3.4-羟基-2-(4-甲氧基苄基)-2H-1,2-苯并噻嗪-3-羧酸甲酯-1,1-二氧化物(4a)的合成。向一150mL三口烧瓶中加入5g化合物3(0.019mol)、30mLDMF及13g碳酸钾(0.095mol),冰浴条件下磁力搅拌反应0.5h后,分2批加入3.2 g4- 甲氧基氯化苄(0.020mol),常温继续反应 2h,TLC[V(石油醚):V(乙酸乙酯)=3:1为展开剂]检测反应结束后,抽滤,向滤液中加入50mL水,用乙酸乙酯(40mL×3)萃取,合并乙酸乙酯层,有机层用水(50mL×3)洗涤,无水硫酸钠干燥,减压浓缩得淡黄色油状物粗品3.9g,收率为52.7%,直接投下一步。
4.4-羟基-2-(4-甲氧基苄基)-N-(3-三氟甲基苯基)-2H-1,2-苯并噻嗪-3-甲酰胺-1,1-二氧化物(5a)的合成。向一100 mL的三口烧瓶中加入3 g化合物4a(0.0077 mol)、1.49 g间三氟甲基苯胺(0.0093 mol)和 30 mL二甲苯,升温至145℃,回流搅拌24 h。TLC[V(石油醚):V(乙酸乙酯)1:1为展开剂]监测反应基本完全。冰盐浴冷却,析出固体,抽滤,丙酮重结晶,干燥,得白色固体 5a(1.5 g),产率:37.4%;m.p.246.7~248.2℃;1H-NMR (400 MHz,DMSO-d6)δ:13.80(s,1H,NH),10.55(s,1H,OH),8.18(s,1H,ArH),8.10(d,J=8.0 Hz,1H,ArH),7.74(t,J=7.6 Hz,2H,ArH),7.69-7.62(m,4H,ArH),7.54(d,J=7.6 Hz,1H,ArH),7.16(s,1H,ArH),6.96(d,J=8.4 Hz,1H,ArH),6.69(d,J=8.4 Hz,1H,ArH),4.52(s,2H,CONH),3.65(s,3H,OCH3);13C-NMR(100 MHz,DMSO-d6)167.4,155.0,138.1,137.1,134.4,132.5,131.0,129.7,129.4,129.0,125.7,125.3,124.8,122.7,122.3,120.9,118.2,111.2,109.5,107.1,56.1,54.5;ESI-MS m/z:503.21(M+-H)。
类似方法合成化合物5b~5f。
5b:白色固体,产率:41.7%,m.p.221.9~223.4℃;1H-NMR(300 MHz,DMSO-d6)δ:13.74(s,1H,NH),10.50(s,1H,OH),8.14(s,1H,ArH),8.09(d,J=8.4 Hz,1H,ArH),7.78~7.72(m,2H,ArH),7.70~7.66(m,1H,ArH),7.63(d,J=7.8 Hz,2H,ArH),7.54(d,J=7.8 Hz,1H,ArH),6.84(d,J=8.1 Hz,2H,ArH),6.74(d,J=7.8 Hz,2H,ArH),4.53(s,2H,CONH),2.04(s,3H,ArCH3);13C-NMR(75MHz,DMSO-d6)167.5,159.4,138.0,137.3,137.0,132.5,129.9,129.7,129.4,129.0,128.2,127.8,125.8,125.4,125.3,122.8,122.7,121.0,118.2,107.3,55.4,20.4;ESI-MSm/z:487.11(M+-H)。
5c:白色固体 ,产率:39.6%,m.p.238.1~240.5℃;1H-NMR (400MHz,DMSO-d6)δ:13.75(s,1H,NH),10.48(s,1H,OH),8.02(d,J=6.8 Hz,1H,ArH),7.75(d,J=7.2 Hz,2H,ArH),7.70(t,J=7.2Hz,1H,ArH),7.63(t,J=7.6Hz,1H,ArH),7.57(d,J=7.6Hz,1H,ArH),7.46(t,J=9.2Hz,1H,ArH),7.01-6.94 (m,5H,ArH),4.58 (s,2H,CONH);13C-NMR(75MHz,DMSO-d6)166.9,138.9,137.3,132.6,132.2,131.6,131.1,130.0,129.7,129.5,129.1,128.6,127.1,126.0,125.0,124.6,122.3,119.9,117.4,106.6,54.2;ESI-MS m/z:507.53(M+-H)。
5d:淡灰色固体,产率:34.2%,m.p.248.6~251.3℃;1H-NMR(400 MHz,DMSO-d6)δ:13.80(s,1H,NH),10.65(s,1H,OH),8.17(s,1H,ArH),8.11(d,J=8.0 Hz,1H,ArH),7.80(d,J=8.0 Hz,2H,ArH),7.73-7.67(m,2H,ArH),7.65-7.59(m,3H,ArH),7.55(t,J=7.6 Hz,1H,ArH),7.30(d,J=8.4 Hz,2H,ArH),4.72 (s,2H,CONH);13C-NMR(75MHz,DMSO-d6)167.2,159.7,146.8,138.7,138.1,136.9,132.7,132.6,131.4,129.7,129.3,125.9,125.4,125.2,123.4,122.9,122.7,122.1,121.0,118.2,118.1,107.2,54.9;ESI-MS m/z:518.17(M+-H)。
5e: 灰 色 固 体 ,产 率 :35.1%,m.p.241.7~243.5℃ ;1H-NMR(300 MHz,DMSO-d6)δ:13.86(s,1H,NH),12.05(s,1H,OH),8.64(d,J=4.5 Hz,1H,PyH),8.25(s,1H,PyH),8.06(d,J=8.1 Hz,1H,PyH),7.99~7.96(m,1H,PyH),7.91~7.86(m,3H,ArH),7.80~7.74(m,1H,ArH),7.70(d,J=8.1 Hz,1H,ArH),7.57(d,J=7.5 Hz,1H,ArH),7.40~7.33(m,2H,ArH),4.65(s,2H,CONH);13C-NMR:(75MHz,DMSO-d6)δ:167.2,159.4,138.8,137.1,132.6,132.4,131.2,130.8,130.5,129.9,128.0,127.8,127.4,127.3,125.7,124.4,122.7,121.2,120.6,107.2,55.4;ESI-MSm/z:476.13(M++H)。
5f:淡黄色固体,产率:30.7%,m.p.227.5~228.9℃;1H-NMR(300MHz,DMSO-d6)δ:14.55(s,1H,NH),7.72-7.62(m,4H,ArH),7.32(s,1H,ThH),6.72(d,J=8.1 Hz,2H,ArH),6.67(d,J=8.1Hz,2H,ArH),4.71(s,2H,CONH),2.34(s,3H,ThCH3),2.06 (s,3H,ArCH3);13C-NMR(75MHz,DMSO-d6)168.4,157.4,136.9,136.7,131.9,131.2,130.4,129.6,129.4,128.9,128.7,127.9,127.5,125.7,125.6,123.7,121.7,110.4,52.8,20.5,11.8;ESI-MSm/z:440.21(M+-H)。
(三)体外抗肿瘤活性测试。该实验以吉非替尼为阳性对照,采用MTT法测试化合物5a~5f的体外抗肿瘤活性:取对数生长期的A549、A431细胞以2×104个/mL细胞密度接种于96孔培养板,100μL/孔,每种细胞各种4块板,培养24h,分别加入 0、0.5、1、2、4、8、16、32、64、128、256、512 及1024μmol/L的待测药物和阳性对照及空白对照200μL,每组设6个复孔,放入CO2培养箱继续培养24h。然后吸弃上清,加入0.5mg/mLMTT的培养液20μL/孔,继续孵育4h。终止培养,吸去孔内培养液,加入200μL/孔二甲基亚砜,振荡10min,使结晶物充分溶解,将试剂对照调零,用全自动酶标仪测定570nm处各孔的OD值。实验重复3次,取平均值。以公式IR=(1-药物组OD值/细胞对照组OD值)×100%计算药物对细胞的抑制率,同时计算IC50值[12]。
二、结果与讨论
(一)化合物的合成。糖精钠和氯乙酸乙酯在高温下经亲核取代反应生成化合物2,以80%乙醇重结晶,产率及纯度较高。化合物2在乙醇钠作用下经Gabriel-Colman反应制得化合物3,在反应过程中要随时监控反应体系的状态,当反应体系过于粘稠时加入少许无水乙醇,保证反应体系为橙黄色浆状物,且此步反应要严格控制无水,TLC监测反应结束后,一定要等温度降至10℃以下,再一次性快速加入浓盐酸和碎冰混合物,否则影响产率,甚至无固体生成。此步反应温度也是能否合成化合物3的关键,文献报道,反应温度始终控制在60℃,但实际操作产率较低,后期依据其他相关文献,先将温度控制在90℃反应0.5h,然后再调至60℃,收率明显提高。原因可能为前期高温给Gabriel-Colman反应过程中的三元环过渡态提供了较高的能量,促使反应快速进行。化合物3与不同苄氯经取代反应制取4a~4d,4a~4d与不同取代氨经酯的氨解反应制得化合物5a~5f,其中5f的产率相对较低,可能由于噻唑氨的活性较苯胺活性低。
(二)体外抗肿瘤活性。6个目标化合物的体外MTT实验结果见表1,从表中数据可知,6个化合物对A431、A549的抑制活性均明显优于美洛昔康,侧链为CF3的化合物苄基取代为吸电子基化合物对A431、A549的抑制活性优于苄基取代为给电子基化合物。化合物5b对A431的抑制活性与吉非替尼相当,对A549的抑制活性优于吉非替尼,化合物5a对A431、A549的抑制活与吉非替尼相当。本课题组准备下一步对侧链为CF3的化合物苄基取代为给电子基化合物的活性和构效关系进一步研究,并对该类化合物的作用机制进行研究。
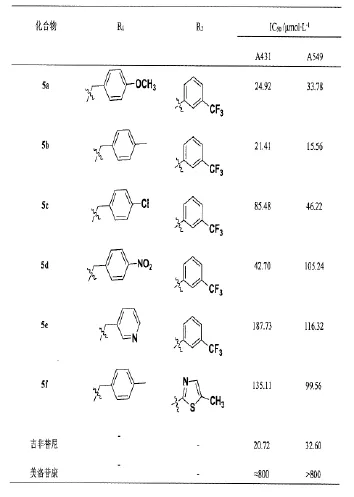
表1 目标化合物的抗肿瘤细胞增殖活性
三、结论
依昔康类药物为模型化合物,运用生物电子等排原理和骨架跃迁原理,设计并合成了6个全新昔康衍生物,其结构均经1H-NMR、13C-NMR、MS谱图确证。MTT实验结果显示,所有化合物对A431、A549均具有一定的抑制活性,其中化合物5b对A549具有显著的抑制活性,IC50值为15.56 μmol?L-1。
参考文献:
[1]ChenW,ZhengR,ZengH,etal.Annualreporton status of cancer in China,2011[J].Chinese Journal of Cancer Research,2015(1):2-12.
[2]陈万青,郑荣寿,张思维,等.2013年中国恶性肿瘤发病和死亡分析[J].中国肿瘤,2016(1):1-8.
[3]Mccubrey JA,Abrams SL,Stadelman K,et al.Targeting signal transduction pathways to eliminate chemotherapeutic drug resistance and cancer stem cells[J].Adv Enzyme Regul,2010(1):285-307.
[4]王丽鸿.分子靶向抗癌药物的研究进展[J].牡丹江医学院学报,2013,34(2):81-83.
[5]Banerjee R,Chakraborty H,Sarkar M.Photophysical studies of oxicam group of NSAIDs:piroxicam,meloxicam and tenoxicam[J].Spectrochim Acta A Mol Biomol Spectrosc,2003(6):1213-1222.
[6]罗维楠,杨俊卿,姜蓉,等.美洛昔康对全脑缺血/再灌注大鼠脑损伤的作用观察 [J].中国药理学通报,2010(11):1455-1459.
[7]辛兵,吉凯强.非甾体类消炎药对卵巢癌抑制作用的研究[J].中华肿瘤防治杂志,2015(16):1266-1269.
[8]Thimmaraju MK,Bheemanapally K,Dharavath R,et al.Improved Anticancer Activity of Meloxicam Hydrogels in K562 andHL60CellLines[J].JYoungPharm,2017(2):209-213.
[9]王杰,许勤龙,李家明,等.1,2-苯并噻嗪类化合物的设计、合成及抗肿瘤活性研究 [J].有机化学,2014(10):2040-2046.
[10]Balssa F,Bonnaire Y.Synthesis of deuterium-labelled meloxicamandpiroxicam[J].JLabelledCompRadiopharm,2007(3):207-210.
[11]吴范宏,王葆丹,奚关根.美洛昔康和炎痛喜康的合成[J].华东理工大学学报自然科学版,2002(3):282-284.
[12]王杰,邬皓,李家明,等.水杨酰芳胺类化合物的设计、合成及体外抗肿瘤活性研究[J].有机化学,2013(5):1026-1034.
