组蛋白H3(Ser10) 磷酸化在维持肿瘤细胞恶性表型中的作用
2018-04-09黎青叶柳晓玲吴小嫩陈丽萍
黎青叶,柳晓玲,吴小嫩,郭 萍 ,陈 雯,陈丽萍*
(中山大学公共卫生学院预防医学系,广东 广州 510080)
2014年世界癌症报告显示,全球每年癌症新发病例约1 400万,死亡约800万,其中肝癌和肺癌的发病率在全球高居前3位;在肝、食道、胃和肺4种脏器的恶性肿瘤中,中国新增病例和死亡人数均居世界首位[1]。肿瘤的形成和发展是多阶段、多因素的过程,其中涉及癌基因的激活和抑癌基因的失活,以及遗传和表观遗传机制的异常[2]。表观遗传是指不涉及DNA序列改变,而在基因表达层面上发生了可遗传的改变,包括DNA甲基化、组蛋白修饰、染色质重塑和非编码RNA[3]。近来肿瘤形成和发展相关的组蛋白修饰日益受到表观遗传学研究学者的重视,且越来越多的结果表明组蛋白修饰在肿瘤发生发展过程中发挥重要的作用[4-5]。
组蛋白H3磷酸化作为有丝分裂的中期相标志,在G1期调控转录活性,在G2/M期可影响染色质凝集[6],具有调控基因转录、DNA损伤修复、细胞分化和凋亡等功能[7-8]。其中,组蛋白H3丝氨酸10位的磷酸化修饰[H3(Ser10)]与细胞周期及基因转录调控等密切相关,以往研究发现组蛋白H3(Ser10)磷酸化作为Aurora B激酶高表达的结果可能参与致癌过程[9];也可通过调节14-3-3ζ蛋白、丝裂素活化蛋白激酶磷酸酶-1(MKP1)、丝裂原和应激激活蛋白激酶1(MSK1)的表达,在染色质结构的调节中发挥作用[10]。多种肿瘤促进因子如表皮生长因子(epidermal growth factor,EGF)诱导组蛋白H3(Ser10)的磷酸化进而激活转录因子连接蛋白1(adaptor protein 1,AP-1)[11]、镉通过下调载脂蛋白E(apolipoprotein E,ApoE)[12]或 改变细胞周期[13]等,在细胞恶性转化中起重要作用。然而,目前对于组蛋白H3(Ser10)磷酸化参与恶性肿瘤细胞表型维持或肿瘤形成的机制仍不明确。
本实验室前期研究发现,在化学致癌物诱导转化细胞模型和肿瘤中,蛋白磷酸酶2A(PP2A)的调控蛋白α4呈高表达[14]。基于以上有关组蛋白H3(Ser10)磷酸化的研究,本文拟探讨组蛋白H3(Ser10)磷酸化在维持肿瘤细胞恶性表型中的作用,并通过α4的表达调控来阐明H3(Ser10)磷酸化在其中的可能机制。
1 材料与方法
1.1 主要试剂
DMEM和RPMI-1640基础培养基、100×青/链霉素、胎牛血清均购于美国Gibco公司;胰蛋白酶购于美国Amresco公司。兔抗人组蛋白H3及Ser10磷酸化多克隆抗体购于美国Cell Signaling Technology公司,兔抗人IGBP1多克隆抗体购于美国Novus Biologicals公司,荧光标记抗兔二抗购于美国Thermo Fisher Scientific公司,鼠抗人β-actin单克隆抗体、辣根过氧化物酶标记的山羊抗小鼠及兔IgG(二抗)购于美国Santa Cruz公司。胸苷(Thymidine)、诺卡达唑(Nocodazole)均购自美国Sigma公司;碘化丙啶(PI)、DAPI购于上海碧云天公司;甲醛、乙醇、氯化镉(CdCl2)均购于广州化学试剂公司;双荧光素酶报告检测试剂盒购于美国Promega公司。
1.2 主要仪器
电泳仪、电泳槽和转膜仪均购于美国Life公司;倒置显微镜购于日本Nikon公司;流式细胞仪购于美国Beckman Coulter公司。
1.3 质粒构建
利用质粒扩增突变试剂盒(Stratagene,美国)构建组蛋白H3(Ser10)位点突变质粒(pBABE-HAH3S10A)。另外以人cDNA为模板,用高保真PCR酶(Thermo Scientific,美国)扩增得到HA-AP1产物后进行凝胶回收,经酶切后装载至pBABE-puro质粒中,构建AP-1 过表达质粒(pBABE-HA-AP1)。α4启动子质粒(p G L 3-α 4)人 α 4启 动 子 片 段 从 h t t p://switchgeargenomics.com获取,设计引物扩增后酶切插入pGL3-MCS-control质粒(中山大学生命科学院庄诗美教授馈赠),所有质粒经测序确认。
1.4 细胞培养
人肝正常细胞株L02来自深圳疾病预防控制中心,人肝癌细胞株Bel7402、HepG2、SMMC-7721和人肺癌细胞株A549来自广州市疾病预防控制中心毒理科。L02、Bel7402、HepG2、SMMC-7721和A549细胞培养于RPMI-1640完全培养基(含10%胎牛血清),置于CO2体积分数为5%、37 ℃恒温箱中培养,待细胞生长至汇合度约为80%时,用0.25%胰蛋白酶消化传代。
1.5 构建组蛋白H3(Ser10)位点突变和AP-1过表达的细胞株
pBABE-HA-H3S10A或pBABE-HA-AP1质粒、包装质粒pCL-ampho按1∶1比例经磷酸钙法转染293FT细胞,产生的病毒经过滤后感染靶细胞Bel7402和A549,经嘌呤霉素(1 μg/mL)筛选获得阳性细胞,并经免疫印迹验证目的基因或标签蛋白HA的表达。
1.6 流式细胞术检测细胞周期
将细胞按每孔1×105~5×105个细胞接种到6 cm培养皿,贴壁24 h后,待细胞生长至汇合度70%~80%时,采用胸苷嘧啶核苷双阻断法的处理组,在细胞培养基中加入2 mmol/L胸苷处理24 h,使细胞处于G1/S期;采用诺考达唑阻抑法的处理组,用100 ng/mL诺考达唑处理24 h使细胞处于G2/M期;对照组不做任何处理。周期阻滞后,消化收集细胞,离心收集的细胞用预冷PBS洗1次,再重悬于500 μL PBS,轻柔震荡,并逐滴添加1.5 mL预冷的纯乙醇,置-20 ℃过夜(至少12 h);上机前在4 ℃,600 g离心10 min处理样品,然后用冰PBS洗1次,静置5 min后再次离心,重悬于300~500 μL的PI/Triton X-100染色液中,37 ℃孵育15 min,后经200目滤网过滤到流式管中,置冰上避光,24 h后用流式细胞仪进行检测;检测结果用Flowjo 7.6软件拟合分析。实验重复3次。
1.7 免疫荧光检测磷酸化H3(Ser10)的表达
将L02细胞按每孔1×104个接种于24孔板,培养24 h后用周期药物阻滞(如1.6中所述)。分别收集对照组、胸苷处理组和诺考达唑处理组的细胞,弃培养基,用预温的PBS洗3次,然后用3.7%的甲醛(用37 ℃预热PBS配制)室温固定15 min;去除固定液,加入PBS 轻柔摇动3 min,重复3次,然后加入含有0.1% Triton X-100的通透液(PBS配制)室温通透5 min。吸走通透液后再用PBS洗3次,加入封闭液(含3% FBS的PBS),在37℃封闭30 min。封闭结束后,加入用封闭液稀释好的一抗p-H3(Ser10)(1∶1 000),4 ℃孵育过夜。次日用PBS洗3次,每次5 min,加入荧光标记Rabbit二抗(1∶1 000),37 ℃孵育1 h。二抗孵育结束后,用PBS洗3次,再用DAPI(1∶1 000)衬染细胞核5 min,PBS洗1次,在免疫荧光显微镜下观察,并拍照记录。实验重复3次。
1.8 蛋白印迹检测H3(Ser10)磷酸化水平改变对α4表达的影响
按上述方法用100 ng/mL诺考达唑将人正常肝细胞株L02和肝癌细胞株HepG2、Bel7402、SMMC-7721阻滞在M2期。收集细胞后使用SDS上样缓冲液直接裂解法获得细胞总蛋白,并使用细胞破碎仪(功率30%,超声5 s,停顿1 s)处理样品,8%~16%梯度胶分离蛋白,湿转法进行转膜。5%的脱脂牛奶封闭1 h,然后分别加入兔抗人抗体α4(1∶5 000)、p-H3(Ser10)(1∶1 000)4℃孵育过夜,次日用PBS/T洗3次,每次5 min,再置于含山羊抗兔IgG HRP(1∶10 000),室温孵育1 h。加上ECL发光剂进入暗室使用X光胶片曝光成像。实验重复3次。
1.9 软琼脂试验检测肿瘤细胞克隆形成能力
构建p-H3(Ser10)低表达的Bel-H3S10A(S10A)细胞株,转染空载质粒的Bel-Vector(AP-1)细胞株为对照。使用DMEM培养基配置底层琼脂(含0.6%琼脂、10%胎牛血清、1%青/链霉素、1%两性霉素B)铺于6孔板冷却备用。消化细胞并计数,按每孔2×104个细胞的密度混悬于顶层琼脂(含0.4%琼脂、10%胎牛血清、1%青/链霉素、1%两性霉素B)中,铺于底层琼脂上,冷却凝固后置于细胞培养箱培养。连续培养21 d后,显微镜下观察克隆并计数。实验重复3次。
1.1 0 双荧光素酶报告试验检测p-H3(Ser10)对α4的转录调控作用
将A549细胞按每孔1×104个的密度接种于96 孔板,贴壁24 h后,按说明书使用Lipofectamine®2000 转染试剂进行pGL3-α4及对照质粒的转染,48 h后用20 μmol/L氯化镉染毒,12 h后使用荧光素酶报告试剂盒上机检测并记录数据。每个样品做3个重复孔,取均值;以质粒pRL-TK海肾光的读取值为内参,计算得到结果。实验重复3次。
1.1 1 统计学方法
应用SPSS 21.0及Graph-pad Prism- 5.0统计学软件处理和分析实验数据。计量资料采用x±s表示,多组间比较采用方差分析,组内两两比较采用LSD-t检验,以p<0.05为差异有统计学意义。
2 结 果
2.1 磷酸化修饰的组蛋白H3(Ser10)在细胞中呈周期性表达
组蛋白H3磷酸化修饰具有周期性,且是细胞有丝分裂中期标志[15],因此首先对L02细胞进行了细胞周期阻滞来检测p-H3(Ser10)的表达。如图1A所示,L02细胞经2 mmol/L胸苷处理24 h(T)后,大多数细胞处于G1和S期,而经100 ng/mL诺考达唑处理24 h(N)后则处于G2/M期。免疫荧光和免疫印迹结果显示(1B和1C),与未处理组相比,p-H3(Ser10)表达水平在胸苷处理后明显下降达71%(p<0.05),几乎无表达,而在诺考达唑处理后表达明显增加,为对照组的3.3倍(p<0.05)。表明,p-H3(Ser10)在细胞中呈周期性表达,且在细胞分裂中期达到最高值。
2.2 磷酸化组蛋白H3(Ser10)在肿瘤细胞中的表达及功能
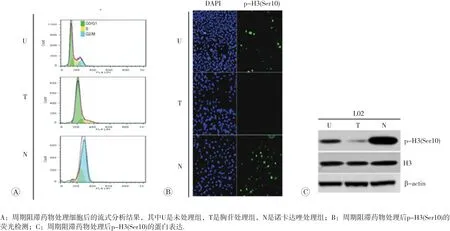
图1 p-H3(Ser10)的周期性表达
将细胞经诺考达唑阻滞在M2期后,采用免疫印迹法检测人正常肝细胞株及肝癌细胞株中p-H3(Ser10)的蛋白表达。结果如图2A所示,与对照L02细胞相比,肝癌细胞株HepG2、Bel7402、SMMC-7721中的p-H3(Ser10)水平升高了(2.61±0.45)倍(p<0.05),但总的组蛋白水平未见明显改变,提示p-H3(Ser10)高表达在肿瘤发生过程中起作用。为验证p-H3(Ser10)在肿瘤中的作用,我们进一步通过对Ser10位点进行突变(丝氨酸突变为丙氨酸),构建了p-H3(Ser10)低表达的Bel-H3S10A细胞株,并对该细胞株的恶性转化功能进行检测。如图2B所示,Ser10位点突变引起的p-H3(Ser10)水平下降,使Bel7402肿瘤细胞株在软琼脂上形成的克隆数目减少30%(p<0.05),表明p-H3(Ser10)在维持肿瘤细胞恶性表型具有重要作用。

图2 p-H3(Ser10)在肿瘤细胞中的表达及功能
2.3 镉诱导组蛋白H3(Ser10)磷酸化修饰增加
进一步探讨p-H3S10过表达参与肿瘤发生的可能机制。和前期实验室结果一致[14],图2A所示,在癌细胞株中除p-H3(Ser10)高表达外,还观察到蛋白磷酸酶2A(PP2A)的调控因子α4的高表达,进一步用氯化镉处理A549细胞。免疫印迹结果如图3所示,在不同浓度的氯化镉(0、10、20和40 μmol/L)处理后,随着氯化镉浓度的增高,p-H3(Ser10)的表达水平明显增加,20和40 μmol/L的浓度处理下分别上升了57%和142%(p<0.05),且具有剂量-反应关系,但总的组蛋白表达水平未见明显改变;同时α4的表达也随着浓度增高而明显增加,分别是对照组的1.41、2.80和2.84倍(p<0.05)。在p-H3(Ser10)诱导激活表达的同时也观察到α4的表达增加,因此我们推测在癌细胞中p-H3(Ser10)可能参与调控α4的表达。
2.4 组蛋白H3(Ser10)磷酸化调控α4的表达
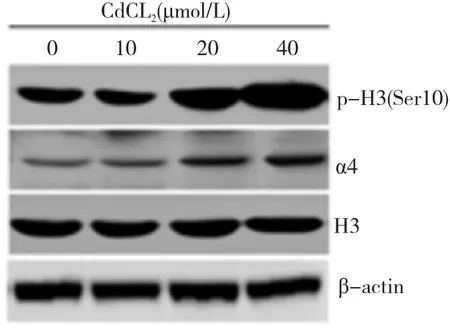
图3 不同浓度镉诱导p-H3(Ser10)和α4表达增加
以往研究表明组蛋白磷酸化可激活转录因子AP-1对靶基因的转录调控作用[16],且通过生物信息学网站预测,α4启动子区有多个AP-1的结合位点(图4A)。为验证p-H3(Ser10)是否通过AP-1调控α4的表达,我们在A549细胞中构建高表达AP-1同时H3(Ser10)位点突变的细胞株A549-AP1-Vector(AP1)和A549-AP1-H3S10A(S10A)。 用20 μmol/L的氯化镉染毒后,检测α4的表达水平。如图4B所示,双荧光素酶报告试验结果显示,AP1细胞经镉处理后,AP-1的转录活性升高了80%(p<0.05);经镉处理后,p-H3(Ser10)低水平表达的S10A细胞株中AP-1的转录活性升高幅度比AP1细胞下降了30%(p<0.05)。一致的是,蛋白印迹结果也显示(图4C),在镉作用下,与高表达AP-1的对照细胞AP1相比,组蛋白H3(Ser10)位点突变株S10A细胞中,p-H3 (Ser10)的表达水平降低51%,同时检测到S10A细胞中的α4蛋白表达较对照细胞株下降47%(p<0.05),但总的组蛋白表达水平未见明显改变。以上结果提示p-H3(Ser10)可通过激活转录因子AP-1调控α4的表达。

图4 p-H3(Ser10)调控α4的表达
3 讨论
组蛋白修饰在转录后水平调节细胞内基因的表达,单个基因位点或全基因组中组蛋白修饰调节模式的紊乱会导致肿瘤的形成[17]。本研究发现H3(Ser10)的磷酸化水平在肝癌细胞株中升高,推测高水平磷酸化修饰的H3(Ser10)具有癌基因功能,而进一步的功能实验则发现p-H3(Ser10)可通过激活转录因子AP-1调控α4的表达来参与肿瘤的发生发展。我们的研究揭示了一条组蛋白异常修饰参与肿瘤发生过程的新信号通路。
以往研究发现H3(Ser10)的磷酸化水平在有丝分裂原和癌基因诱导转化的小鼠成纤维母细胞中是增加的[18],并参与EB病毒潜在膜蛋白-1(LMP1)诱导的鼻咽癌形成过程[19]。相一致的是,我们也发现全基因组水平的组蛋白H3磷酸化修饰表达升高,这与组蛋白H3(Ser10)位磷酸化水平在以上肿瘤中升高的报道是一致的,证实其很可能参与了肿瘤形成的过程。此外,也有研究证实H3(Ser10)的磷酸化修饰与快速反应基因(immediate-early gene,IE)相关,包括原癌基因c-fos和c-jun[20]。IE基因反应在分化、有丝分裂、疾病如癫痫和癌症过程中发挥作用[21-22]。
本研究发现H3(Ser10)的磷酸化水平随着细胞周期呈现规律性变化,这和以往的研究一致[23]。H3(Ser10)的磷酸化水平在癌细胞株中升高,也在可诱导细胞恶性转化的化学致癌物氯化镉诱导下升高,这与α4的表达相似且趋势一致。我们之前发现PP2A的调节蛋白α4在人恶性细胞转化过程中起着重要的调控作用,α4在转化细胞中表达显著上调并且在肺腺癌组织中呈高表达水平。本研究中α4在肝癌细胞株HepG2、Bel7402、SMMC-7721中的表达水平较L02细胞均显著升高,且氯化镉染毒处理也可以诱导α4的表达上调,进一步支持了α4在化学致癌和恶性转化表型中的重要作用。α4基因启动子存在转录因子AP-1的调控区域,我们构建了高表达AP-1同时H3(Ser10)位点突变的细胞株,发现转录因子AP-1对α4的表达调控作用减弱,与高表达AP-1的对照细胞相比,H3(Ser10)位点突变细胞的p-H3 (Ser10)和α4表达水平均下降近50%;且在细胞受到致癌化学物氯化镉的刺激下,低表达p-H3(Ser10)细胞的α4表达升高幅度比对照细胞下降近50%,说明p-H3(Ser10)可通过激活转录因子AP-1调控α4的表达,提示组蛋白H3(Ser10)磷酸化参与了化学物致癌过程中α4的调控通路。
综上,本研究发现组蛋白H3(Ser10)磷酸化修饰在肝肿瘤细胞中呈过表达水平,且在化学致癌物诱导过程中显著上调,提示组蛋白H3(Ser10)参与肿瘤细胞维持恶性表型的过程,其机制可能是组蛋白H3(Ser10)通过转录因子AP-1调控促癌因子α4的表达。因此,我们认为组蛋白H3(Ser10)磷酸化有望作为癌症治疗的一个新靶点。
[1] STEWART B W,WILD C P. World Cancer Report 2014[M].IARC,2014.
[2] JOHNSON C,WARMOES M O,SHEN X,et al. Epigenetics and cancer metabolism[J]. Cancer Lett,2015,2 Sup A(356):309-314.
[3] MOSHE S. Nongenetic inheritance and transgenerational epigenetics[J]. Trends Mol Med,2015,21(2):134-144.
[4] LI D,BI F F,CAO J M,et al. Poly (ADP-ribose) polymerase 1 transcriptional regulation:A novel crosstalk between histone modification H3K9ac and ETS1 motif hypomethylation in BRCA1-mutated ovarian cancer[J]. Oncotarget,2014,1(5):291-297.
[5] SUN Y,BELL J L,CARTER D,et al. WDR5 supports an NMyc transcriptional complex that drives a protumorigenic gene expression signature in neuroblastoma[J]. Cancer Res,2015,75(23):5143-5154.
[6] NOWAK S J,CORCES V G. Phosphorylation of histone H3:a balancing act between chromosome condensation and transcriptional activation[J]. Trends Genet,2004,4(20):214-220.
[7] METZGER E, YIN N A, WISSMANN M, et al.Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation[J]. Nat Cell Biol,2007,10(1):53-60.
[8] YOSHIDA I,IBUKI Y. Formaldehyde-induced histone H3 phosphorylation via JNK and the expression of protooncogenes[J]. Mutat Res,2014(770):9-18.
[9] OTA T,SUTO S,KATAYAMA H,et al. Increased mitotic phosphorylation of histone H3 attributable to AIM-1/Aurora-B overexpression contributes to chromosome number instability[J].Cancer Res,2002,62(18):5168-5177.
[10] SHARMA A K,MANSUKHA,VARMA A,et al. Molecular modeling of differentially phosphorylated serine 10 and acetylated lysine 9/14 of histone H3 regulates their interactions with 14-3-3 zeta, MSK1, and MKP1[J]. Bioinform Biol Insights,2013(7):271-288.
[11] CHOI H S,CHOI B Y,CHO Y Y,et al. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation[J]. Cancer Res,2005,65(13):5818-5827.
[12] TAKEDA S M,TERAOKA-NISHITANI N,YAMAGATA A,et al. Cadmium-induced malignant transformation of rat liver cells:Potential key role and regulatory mechanism of altered apolipoprotein E expression in enhanced invasiveness[J].Toxicology,2017,382:16-23.
[13] NGALAME N N,WAALKES M P,TOKAR E J. Silencing KRAS overexpression in cadmium-transformed prostate epithelial cells mitigates malignant phenotype[J]. Chem Res Toxicol,2016,9(29):1458-1467.
[14] CHEN L P,LAI Y D,LI D C,et al. [alpha]4 is highly expressed in carcinogen-transformed human cells and primary human cancers[J]. Oncogene,2011,30(26):2943.
[15] XU Y M,Du J Y,LAU A T. Posttranslational modifications of human histone H3:an update[J]. Proteomics,2014,17/18(14):2047-2060.
[16] SCHMUCKER A C,WRIGHT J B,COLE M D,et al. Distal interleukin-1 beta (IL-1 beta) response element of human matrix metalloproteinase-13 (MMP-13) binds activator protein 1 (AP-1)transcription factors and regulates gene expression[J]. J Biol Chem,2012,2(287):1189-1197.
[17] VARDABASSO C,HASSON D,RATNAKUMAR K,et al.Histone variants:emerging players in cancer biology[J]. Cell Mol Life Sci,2014,3(71):379-404.
[18] STRELKOV I S,DAVIE J R. Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogenetransformed mouse fibroblasts[J]. Cancer Res,2002,62(1):75-78.
[19] LI B, HUANG G, ZHANG X, et al. Increased phosphorylation of histone H3 at serine 10 is involved in Epstein-Barr virus latent membrane protein-1-induced carcinogenesis of nasopharyngeal carcinoma[J]. BMC Cancer,2013,13:124.
[20] HAZZALIN C A,BARRATT M J,MAHADEVAN L C,et al.The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades:MSK1 as a potential histone H3/HMG-14 kinase[J]. EMBO J,1999,18(17):4779-4793.
[21] HERSCHMAN H R. Primary response genes induced by growth factors and tumor promoters[J]. Annu Rev Biochem,1991(60):281-319.
[22] MCMAHON S B,MONROE J G. Role of primary response genes in generating cellular responses to growth factors[J].FASEB,1992,6(9):2707.
[23] FEHRI L F,RECHNER C,JANSSEN S,et al. Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation[J]. Epigenetics,2009,8(4):577-586.
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
