Melastatin-related transient receptor potential 2 channel in Aβ42-induced neuroin flammation: implications to Alzheimer’s disease mechanism and development of therapeutics
2018-04-04LinyuWei,SharifahAlawieyahSyedMortadza,Lin-HuaJiang
Alzheimer’s disease (AD) is an age-related neurodegenerative disease that represents the most common cause of dementia among the elderly people. With the increasingly aging population, AD has presented an overwhelming healthcare challenge to modern society; the World Alzheimer Report 2015 has estimated that 46.8 million people worldwide lived with dementia in 2015 and this number will rise to 74.7 million in 2030 and that the total cost of dementia was 818 billion in US$ in 2015 and will reach two trillion in 2030. Post-mortem studies have identi fied two histopathological hallmarks in the brains of AD patients; extracellular senile plaque with elevated deposition of amyloid β (Aβ) peptides, and intracellular neuro fibrillary tangle composed of hyper-phosphorylated microtubule-associated protein tau.Etiologically, progressive neuronal loss within the cerebral cortex and hippocampus regions of the brain leads to irreversible decline in, and eventually complete loss of, memory and other cognitive functions that afflict AD patients. The widely-accepted amyloid cascade hypothesis for AD pathogenesis holds that accumulation and aggregation of neurotoxic Aβ peptides, due to imbalance of their generation and clearance as a result of changes in genetic makeup, aging and/or exposure to environmental risk factors, is a major and early trigger of AD. This hypothesis has continuously gained support by preclinical and clinical studies (Selkoe and Hardy, 2016). However, the intensive and costly drug discovery efforts over the past decades based on such a hypothesis have proved extremely frustrating in developing effective therapeutics to treat or slow down the progress of AD, highlighting the need for more research to improve our understanding towards the cellular and molecular mechanisms by which Aβ peptides bring about neurotoxicity and cognitive dysfunction.
Neuro-inflammatory responses or changes have been well documented since early studies examining the brains of AD patients and been replicated in the brains of rodent models of AD. Microglial cells,derived from yolk sac during early development, are the major immunocompetent cells resident in the central nervous system. These cells therefore have the sensor and effector functions and exhibit strong phagocytic capacity, like macrophage cells in the peripheral immune system. Under healthy conditions or in a resting state, microglial cells adopt a rami fied morphology and act as a sentinel to survey the brain parenchyma for pathogens and cellular debris and remove them mainly via phagocytosis (Wolf et al., 2017). In response to damage and infection or in contact with damage/pathogen-associated molecular patterns (DAMP/PAMP) molecules, microglial cells become activated with changing to an amoeboid morphology and initiate immune responses by generating proin flammatory mediators such as cytokines, chemokines and reactive oxygen species (ROS) in order to restore the normal brain homeostasis. There is accumulating evidence to show dual and opposing roles of microglial cells in AD. Microglial cells on one hand play an active role in clearing Aβ by generation of anti-Aβ antibodies and phagocytosis. Such a bene ficial role however becomes progressively weakened and eventually inefficient with aging or following exposure to excessive Aβ peptides. On the other hand,accumulation of Aβ peptides as DAMP molecules can induce microglial activation and dysregulated release of proin flammatory mediators that cause neurotoxicity. Further release of DAMP molecules by degenerating neurons forms a vicious positive feedback loop leading to development of chronic proin flammatory environments that facilitate generation and accumulation of Aβ peptides and are detrimental to neuronal survival. Such Aβ-induced neuroinflammation becomes irreversible, which may offer some explanation why therapeutic strategies aiming to halt the generation of Aβ peptides are ineffective in treating AD in its late stage. There is rapidly rising interest in microglial cells for their role in Aβ-induced neuroin flammation and the underlying mechanisms and, additionally, targeting microglial cell functions as a novel and/or additional avenue of developing therapeutics for AD (Minter et al., 2016; Wes et al., 2016; Ramirez et al., 2017;Sarlus and Heneka, 2017).
Melastatin-related transient receptor potential 2 (TRPM2) channel belongs to the mammalian superfamily of tetrameric transient receptor potential channels, with each subunit containing six transmembrane segments and intracellular N- and C-termini, and functions as a Ca2+-permeable cationic channel that is exclusively gated by intracellular ADP-ribose (ADPR) (Figure 1). TRPM2 channel is highly sensitive to activation by ROS indirectly through stimulating ADPR-generating mechanisms including poly(ADPR) polymerase-1(PARP-1) in the nucleus (Jiang et al., 2010). Exposure of microglial cells to ROS elevated intracellular Ca2+concentrations, and such Ca2+responses were suppressed by genetic knockout of the TRPM2 expression (TRPM2-KO), pharmacological inhibition of the PARP-1 or the TRPM2 channel, or removal of extracellular Ca2+, supporting that the TRPM2 channel in microglial cells acts as a Ca2+-permeable channel on the cell surface (Syed Mortadza et al., 2017). Recent studies have revealed TRPM2 channel as a novel mechanism mediating Aβ-induced neuroin flammation (Ostapchenko et al., 2015; Aminzadeh et al., 2018; Syed Mortadza et al., 2018). The amyloid precursor protein/presenilin 1 (APP/PS1) mice, which express excessive amounts of Aβ and develop amyloid deposition in hippocampus and other brain regions, are a widely-used AD model in study of Aβ-related pathogenesis. Ostapchenko et al have recently shown that TRPM2-KO rescued the APP/PS1 mice from Aβ-induced AD-related synaptic loss in hippocampus, microglial activation, and age-related deficit in memory (Ostapchenko et al., 2015). This study provides the first line of evidence to demonstrate a critical role for the TRPM2 channel in Aβ-induced microglial cell activation and neuroinflammation as well as neurotoxicity. Two more recent studies have shed further light on the role of the TRPM2 channel in microglial cells in Aβ-induced neuroin flammation and the underlying molecular or signalling mechanisms (Aminzadeh et al., 2018; Syed Mortadza et al., 2018).
It has been shown that TRPM2 channel or more specifically TRPM2-mediated Ca2+signalling plays an important role in coupling mitochondrial generation of ROS in response to exposure to DAMP/PAMP molecules such as charged lipids and particulate crystals to activation of the NLRP3 inflammasome and caspase-1 to induce maturation and secretion of interleukin-1β (IL-1β) in macrophage cells (Zhong et al., 2013). Thus, Aminzadeh et al have drawn their attention to a similar question, namely, whether the TRPM2 channel in microglial cells has a role in Aβ-induced generation of IL-1β(Aminzadeh et al., 2018). They showed that exposure to 10 μM Aβ42,the most neurotoxic Aβ peptide, induced generation of ROS through mitochondria and also nicotinamide adenine dinucleotide phosphate(NADPH)-dependent oxidases (NOX), and generation of IL-1β in microglial cells that were prior primed with lipopolysaccharide (LPS).LPS is the major molecular component of the outer membrane of Gram-negative bacteria that is widely used as a PAMP molecule to induce immune cell activation, particularly synthesis of pro-IL-1β,via toll-like receptor 4 (TLR4) (Figure 1). Aβ42-induced generation of IL-1β was strongly inhibited by scavenging of ROS, inhibition of the PARP-1 to prevent ROS-induced activation of the TRPM2 channel,or chelation of intracellular Ca2+. In addition, the study showed that exposure to Aβ42induced activation of caspase-1, which was also suppressed by inhibition of PARP-1 activation or increase in the intracellular Ca2+concentration. These findings have led the authors to conclude that the TRPM2 channel in microglial cells plays a vital role in Aβ42-induced generation of IL-1β via transducing NOX-mediated and/or mitochondrial generation of ROS to an intracellular Ca2+signal that activates the NLRP3 in flammasome and caspase-1 (highlighted in redFigure 1).
We have also investigated the role of the TRPM2 channel in Aβ42-induced neuroinflammation, focusing on microglial cell activation and generation of tumour necrosis factor-α (TNF-α) (Syed Mortadza et al., 2018), another proinflammatory cytokine critically implicated in AD pathogenesis (Minter et al., 2016). Exposure to Aβ42at biologically relevant concentrations (30—300 nM) induced microglial cells to undergo prominent changes in cell morphology from a rami fied and resting state-like morphology to a more activated statelike morphology. In addition, exposure to Aβ42stimulated microglial cells to express and secret TNF-α. Aβ42-induced change in cell morphology or states, expression and secretion of TNF-α were prevented by TRPM2-KO or inhibition of the PARP-1 or TRPM2 channel,revealing a critical role for the TRPM2 channel in Aβ42-induced microglial cell activation and generation of TNF-α. Exposure to Aβ42increased the intracellular Ca2+concentration that was largely prevented by TRPM2-KO, inhibition of the PARP-1 or the TRPM2 channel, or removal of extracellular Ca2+, indicating that TRPM2 channel mediates Aβ42-induced Ca2+signalling via extracellular Ca2+in flux. We further examined the signalling mechanisms responsible for Aβ42-induced activation of the TRPM2 channel. Exposure to Aβ42induced generation of ROS and activation of PARP-1 in the nucleus. Aβ42-induced generation of ROS and activation of the PARP-1 and TRPM2 channel were markedly suppressed by inhibition of protein kinase C (PKC) or NOX, particularly the NOX1/4 and NOX2 isoforms, and also by inhibition of the Ca2+-sensitive tyrosine kinase PYK2 or downstream mitogen-activated protein kinases MEK/ERK. Interestingly, Aβ42-induced activation of the PARP-1 was signi ficantly attenuated but not completely abrogated by TRPM2-KO. Furthermore, Aβ42-induced activation of the PARP-1 in TRPM2-de ficient microglial cells was fully blocked by inhibition of PKC or NOX, but not affected by inhibition of PYK2 or MEK/ERK.Taken together, these observations support the hypothesis that Aβ42-induced PKC/NOX-mediated generation of ROS and ROS-induced generation of ADPR via activation of PARP-1 are vital in the initial activation of the TRPM2 channel and that TRPM2-mediated Ca2+in flux and subsequent activation of the PYK2/MEK/ERK signalling pathway serve as a positive feedback to further enhance activation of the PARP-1 and TRPM2 channel (highlighted in green inFigure 1).
In summary, recent studies have disclosed novel TRPM2 channel mechanisms by which Aβ peptides, more specifically Aβ42, induce microglial cell activation and generation of IL-1β and TNF-α, the two key proinflammatory cytokines in AD-related neuroinflammation(Figure 1). Nonetheless, it is evident that more studies are required to provide unifying and more insights into TRPM2-mediated neuroinflammation and the underlying molecular mechanisms and associated signalling pathways, thereby further improving the amyloid cascade hypothesis. Currently, there is limited availability with respect to TRPM2 specific inhibitors. With a body of compelling evidence accumulated from the above-discussed and many other recent studies that support the critical role of the TRPM2 channel in mediating numerous ROS-related diseases, the TRPM2 channel has become an increasingly attractive drug target and, as a result, there is growing interest in development of selective and potent TRPM2 inhibitors. It will be important and exciting therapeutically to test in the near future when such inhibitors become available whether pharmacological intervention of the TRPM2 channel mechanisms of Aβ-induced neuroin flammation provides a plausible therapeutic approach to mitigate the pathogenesis and progression of AD.
The research work described in this article is supported in parts by grants from the Disciplinary Group of Psychology and Neuroscience Xinxiang Medical University, China (2016PN-KFKT-06), Department of Education of Henan Province, China (16IRTSTHN020), the National Natural Science Foundation of China (31471118), and UK Alzheimer’s Research Trust (ART/PPG2009A/2).
Linyu Wei, Sharifah Alawieyah Syed Mortadza, Lin-Hua Jiang*
Sino-UK Joint Laboratory of Brain Function and Injury of Henan
Province, and Department of Physiology and Neurobiology, Xinxiang Medical University, Xinxiang, Henan Province, China (Wei L, Jiang L-H)
School of Biomedical Sciences, Faculty of Biological Sciences,University of Leeds, Leeds, UK (Wei L, Syed Mortadza SA, Jiang L-H)
orcid:0000-0001-6398-0411 (Lin-Hua Jiang)
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under identical terms.
Open peer reviewer:Maria E. Figueiredo-Pereira, Hunter College, USA.
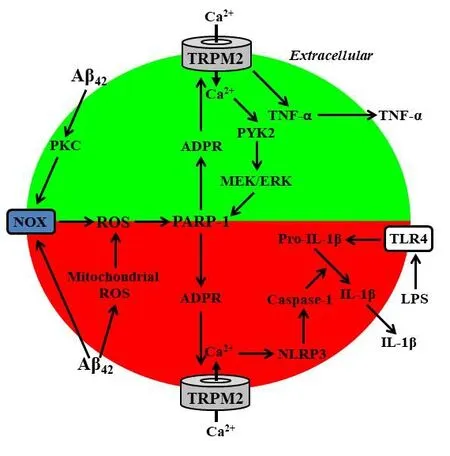
Figure 1 Schematic description of the melastatin-related transient receptor potential 2 (TRPM2) channel mechanisms of amyloid β-induced neuroin flammation.
Aminzadeh M, Roghani M, Sarfallah A, Riazi GH (2018) TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int Immunopharmacol 54:78-85.
Jiang LH, Yang W, Zou J, Beech DJ (2010) TRPM2 channel properties, functions and therapeutic potentials. Expert Opin Ther Targets 14:973-988.
Minter MR, Taylor JM, Crack PJ (2016) The contribution of neuroin flammation to amyloid toxicity in Alzheimer’s disease. J Neurochem 136:457-474.
Ostapchenko VG, Chen M, Guzman MS, Xie YF, Lavine N, Fan J, Beraldo FH,Martyn AC, Belrose JC, Mori Y, MacDonald JF, Prado VF, Prado MA, Jackson MF (2015) The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment. J Neurosci 35:15157-15169.
Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, Lopez-Cuenca I, Rojas P, Trivino A, Ramirez JM (2017) The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and Glaucoma.Front Aging Neurosci 9:214.
Sarlus H, Heneka MT (2017) Microglia in Alzheimer’s disease. J Clin Invest 127:3240-3249.
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595-608.
Syed Mortadza SA, Sim JA, Stacey M, Jiang LH (2017) Signalling mechanisms mediating Zn(2+)-induced TRPM2 channel activation and cell death in microglial cells. Sci Rep 7:45032.
Syed Mortadza SA, Sim JA, Neubrand VE, Jiang LH (2018) A critical role of TRPM2 channel in Abeta42-induced microglial activation and generation of tumor necrosis factor-alpha. Glia 66:562-575.
Wes PD, Sayed FA, Bard F, Gan L (2016) Targeting microglia for the treatment of Alzheimer’s Disease. Glia 64:1710-1732.
Wolf SA, Boddeke HW, Kettenmann H (2017) Microglia in physiology and disease. Annu Rev Physiol 79:619-643.
Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L (2013) TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 4:1611.
杂志排行

中国神经再生研究(英文版)的其它文章
- The biological clock: future of neurological disorders therapy
- Cerebral ischemia and neuroregeneration
- SNARE complex in axonal guidance and neuroregeneration
- Heterozygous carriers of galactocerebrosidase mutations that cause Krabbe disease have impaired microglial function and defective repair of myelin damage
- The relaxin peptide family – potential future hope for neuroprotective therapy? A short review
- Roles of neural regeneration in memory pharmacology