超声浸提-高效液相色谱法测定刺三加叶中绿原酸
2018-03-27高林晓王玉林毛海立杨再波
高林晓,郭 蒙*,王玉林,毛海立,杨再波
(1.黔南民族师范学院化学化工学院,贵州 都匀 558000;2.贵州省普通高校民族药用植物资源开发工程研究中心,贵州 都匀 558000)
刺三加(Acanthopanax trifoliatusL.Merr.)为五加科五加属攀援状灌木,别名有白簕、白簕根、三加皮、三叶五加、刺三甲和簕钩菜等,其嫩芽富含粗纤维和多种微量元素,有改善胃肠功能、清热解毒、防治心脑血管疾病、抗疲劳、减肥等功效[1]。刺三加叶中富含黄酮、酚酸和苯丙素类等物质,其中绿原酸是植物体在有氧呼吸过程中经莽草酸途径产生的一种苯丙素类化合物,具有抗菌、抗病毒、增高白血球、保肝利胆、抗肿瘤、降血压、清除自由基和兴奋中枢神经系统等作用[2]。刺三加茶作为广东恩平特色的农产品已进入人们的视野,一条刺三加产业链已初步形成[3]。
据文献报道,绿原酸含量测定的方法主要有紫外分光光度法[4-5]、高效液相色谱法(high performance liquid chromatography,HPLC)[6-11]、毛细管电泳法[12]、电化学法[13]和伏安法[14]等。这些方法在实际应用中都存在一定的局限。紫外分光光度法不能消除绿原酸同分异构体在同一波长下也存在吸收峰的干扰,结果严重偏高;毛细管电泳法中电渗会因样品组成而变化,进而影响分离重现性,由于毛细管直径小,使光路太短,用一些检测方法时,灵敏度较低;电化学法受干扰物,环境外界影响因素较大。已报道测定刺三加中绿原酸含量的方法有紫外分光光度法[4],但鲜有高效液相色谱法测定刺三加叶中绿原酸含量的方法报道。
本研究采用超声浸提-高效液相色谱法建立快速测定刺三加叶中绿原酸含量的方法,以期为制定科学合理的刺三加质量控制标准提供科学依据。
1 材料与方法
1.1 材料与试剂
实验中所用刺三加叶分别采自贵州省天柱县渡马乡和广东恩平。由黔南民族师范学院生物科学与农学院黄小娜副教授鉴定为五加科五加属植物刺三加叶。
甲醇、乙腈(均为色谱纯):德国Merck公司;乙醇(分析纯):成都金山化学试剂有限公司;石油醚(分析纯):天津市化学试剂供销公司;蒸馏水为实验室自制;绿原酸标准品(纯度为98.09%,批号:LC18522):合肥博美生物科技有限公司。
1.2 仪器与设备
Agilent1260高效液相色谱仪:美国安捷伦科技有限公司;TU-1901双光束紫外可见分光光度计:北京普析通用仪器有限公司;FW177高速万能粉碎机:天津市泰斯特有限公司;SHZ-III型循环水真空泵:郑州英三谷华仪器有限公司;BS110S电子天平:北京赛多利斯仪器系统有限公司;DL-360D智能超声波清洗器:上海之信仪器有限公司;RE52A旋转蒸发仪:上海亚荣生化仪器厂。
1.3 方法
1.3.1 样品处理方法
称取干燥并粉碎的刺三加叶8.00 g,过40目筛,置于250 mL锥形瓶中,加10倍煮沸的蒸馏水浸提3 min,冷却后进行粗滤。再加10倍水同法处理,抽滤,弃去浸提液,得滤渣。加100 mL石油醚于滤渣中脱脂、脱色3 h,重复4次,至石油醚层近无色,抽滤,倾去醚液,样品置于通风处干燥备用。准确称取上述刺三加叶粉末2.000 2 g置150 mL锥形瓶中,准确加入体积分数60%乙醇溶液100 mL,超声间歇提取2次,每次30 min,超声功率100 W,合并提取液,减压蒸干,用体积分数60%乙醇溶液溶解并定容于100 mL棕色容量瓶中,用0.45 μm的有机相微孔滤膜滤过,取续滤液即得供试品溶液[15-16]。
1.3.2 绿原酸检测的色谱条件
采用高效液相色谱法检测刺三加叶中绿原酸的含量。色谱条件:ZORBAX SB-C18色谱柱(4.6 mm×250 mm,5 μm);流动相为甲醇-0.2%磷酸水(15∶85,V/V)溶液;检测波长327 nm;流速1.0 mL/min;进样量10 μL;柱温30℃。
1.3.3 绿原酸标准曲线的绘制
准确称取105℃干燥至质量恒定的绿原酸标准品0.0052g,置于25mL棕色容量瓶中,加入1.0mL甲醇溶解,再加蒸馏水稀释至刻度,摇匀,配制成0.208mg/mL的绿原酸标准储备溶液;精密吸取上述溶液2.0 mL置于100 mL棕色容量瓶中,加蒸馏水稀释至刻度,摇匀,即得绿原酸标准储备液(质量浓度为4.16μg/mL),放置于2~8℃冰箱避光保存。
分别精密吸取0.2 mL、0.5 mL、1.0 mL、2.0 mL、3.0 mL、4.0 mL、5.0 mL、6.0 mL和10.0 mL质量浓度为4.16 μg/mL的绿原酸标准储备液,置于10 mL量瓶中,加蒸馏水稀释至刻度,摇匀,制成质量浓度分别为0.08 μg/mL、0.21 μg/mL、0.42μg/mL、0.83μg/mL、1.25μg/mL、1.66μg/mL、2.08μg/mL、2.50 μg/mL和4.16 μg/mL的标准溶液,所得溶液用0.22 μm有机相微孔滤膜过滤,按照1.3.2色谱条件测定峰面积,并以绿原酸质量浓度(C)为横坐标,对应的色谱峰面积(A)为纵坐标,绘制绿原酸标准曲线。
2 结果与分析
2.1 波长的确定
在波长200~400 nm范围内对一定质量浓度的绿原酸标准溶液进行紫外吸收扫描,结果如图1所示。由图1可知,绿原酸在波长218 nm及327 nm处有最大紫外吸收,考虑到波长218 nm左右紫外吸收有较多物质干扰,同时该波长接近流动相甲醇的紫外截止波长,对样品测定会有一定影响,故选择327 nm作为检测绿原酸的最佳波长。
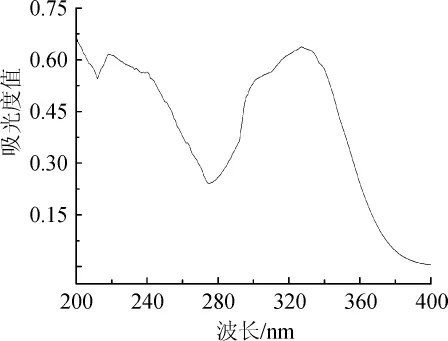
图1 绿原酸标准品紫外扫描图Fig.1 UV scanning graph of chlorogenic acid standard substance
2.2 色谱条件的优化选择
分别考察了甲醇-水-磷酸、乙腈-水-磷酸、甲醇-水-冰醋酸、乙腈-水-冰醋酸和甲醇-水-甲酸等流动相系统[17-21],预实验发现以甲醇-0.2%磷酸水溶液(15∶85,V/V)进行分离的分析效果较好,在该流动相下,刺三加叶中绿原酸的分离度符合要求,具有重现性好、背景噪音干扰小和所得峰形对称性好等优点,所以最终选择甲醇-0.2%磷酸水(15∶85,V/V)溶液为流动相。考察柱温(20℃、25℃、30℃、35℃)、体积流量(0.6 mL/min、0.8 mL/min、1.0 mL/min)对绿原酸的分离效果,结果柱温30℃、体积流量为1.0 mL/min峰型对称性好,理论板数高,分离效果较佳。
2.3 供试品溶液制备方法的优化选择
比较了以甲醇、乙醇和蒸馏水分别作溶剂,使用超声的方法提取,为了防止绿原酸在较高温度条件下降解,超声过程控制温度为20℃。实验发现以蒸馏水作为提取溶剂时有效成分的提取率较低,且杂质较多,以甲醇、乙醇作为提取溶剂时,有效成分的峰形及含量无显著差异,考虑到甲醇的毒性比乙醇大,故选择乙醇为提取溶剂。在此基础上,又对乙醇的体积分数(40%、50%、60%、70%、80%)及超声提取时间(10 min、20 min、30 min、40 min)进行了考察,确定体积分数60%乙醇溶液,20℃超声提取30 min,并旋蒸浓缩,用体积分数60%乙醇溶液溶解定容于100 mL棕色容量瓶中,绿原酸提取最佳。因此本实验选用体积分数60%乙醇溶液超声处理30 min,旋蒸浓缩样品的提取方法。
2.4 绿原酸标准品及样品高效液相色谱分析
按照“1.3.2”项下色谱条件,采用高效液相色谱法分别检测了刺三加叶中提取绿原酸的供试品溶液、绿原酸标准品及体积分数60%乙醇的空白溶液,结果见图2。由图2可知,绿原酸标准品的保留时间为25.653 min,峰形较窄,对称性好。样品提取液中绿原酸的保留时间为25.656 min,与绿原酸标准品的保留时间几乎没有差别,由此可以判定刺三加叶提取液中含有绿原酸。
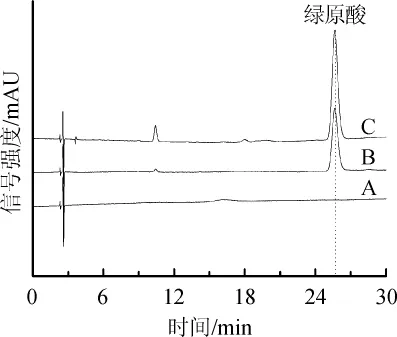
图2 空白(A)、绿原酸标准品(B)和供试品(C)的HPLC色谱图Fig.2 HPLC chromatogram of blank(A),chlorogenic acid standard substance(B)and sample(C)
2.5 绿原酸标准曲线的绘制
采用高效液相色谱法测定绿原酸标准溶液色谱峰面积,以绿原酸质量浓度(C)为横坐标,色谱峰面积(A)为纵坐标,得到绿原酸标准溶液线性回归方程,结果见图3。
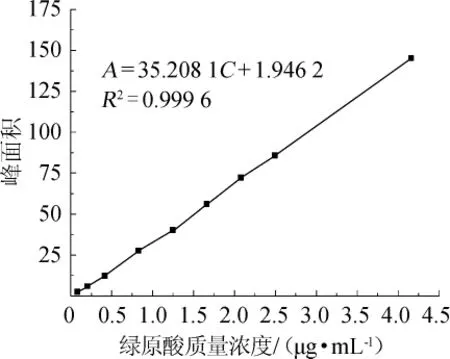
图3 绿原酸标准曲线Fig.3 Standard curve of chlorogenic acid
由图3可知,绿原酸线性回归方程为A=35.2081C+1.9462,相关系数R2=0.9996,表明绿原酸质量浓度在0.08~4.16μg/mL范围内与峰面积呈良好的线性关系,可根据线性回归方程计算刺三加叶中绿原酸的含量。
2.6 方法学考察
2.6.1 精密度试验
准确吸取2.08 μg/mL绿原酸标准储备液,按“1.3.2”节色谱条件连续进样6次,记录绿原酸的峰面积,计算绿原酸含量及相对标准偏差(relative standard deviation,RSD),结果见表1。由表1可知,绿原酸含量的RSD为0.81%,表明仪器精密度良好。

表1 精密度试验结果Table 1 Results of precision experiments
2.6.2 重复性试验
精密移取“1.3.1”节中同一批供试品溶液适量,取样6份,再按“1.3.4”节色谱条件进样测定,记录供试品中绿原酸峰面积,计算绿原酸含量及相对标准偏差RSD,结果见表2。由表2可知,绿原酸含量的RSD为0,表明该方法重复性好。

表2 重复性试验结果Table 2 Results of repeatability experiments
2.6.3 稳定性试验
精密吸取“1.3.1”节中同一批供试品溶液适量,分别于室温条件下放置0、2 h、4 h、8 h、12 h、24 h,按“1.3.2”项下色谱条件进样测定,记录供试品溶液中绿原酸峰面积,计算绿原酸含量及相对标准偏差RSD,结果见表3。由表3可知,绿原酸含量的RSD为0.19%,表明供试品溶液在室温放置24 h内稳定性良好。

表3 稳定性试验结果Table 3 Results of stability experiments
2.6.4 定量限与检测限
按“1.3.2”节中色谱条件,取4.16 μg/mL绿原酸标准储备液,稀释成一系列不同质量浓度溶液进样测定。当信噪比(峰高与基线噪音之比)S/N=10±1时,绿原酸的定量限为0.021 μg/mL;当信噪比S/N=3±1时,绿原酸的检测限为0.007 μg/mL,结果见表4。由表4结果表明,仪器的噪音低,灵敏度高。
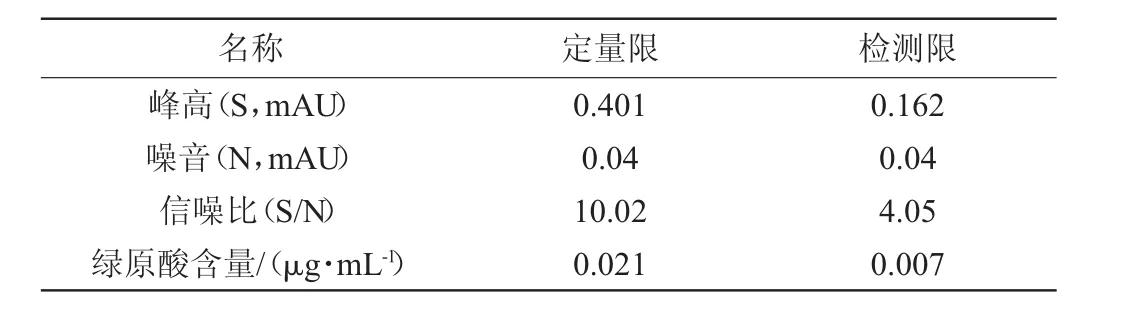
表4 定量限与检测限试验结果Table 4 Experiment results of limits of quantitation and detection
2.6.5 加样回收率试验
按照1.3.1样品处理方法平行制取6份供试品溶液,各精密吸取0.15 mL供试品溶液,用体积分数60%乙醇溶液溶解定容至10 mL棕色容量瓶中,摇匀,从上述6份供试品溶液中各移取0.15mL,绿原酸标准储备液(4.16μg/mL)5mL,置于10 mL棕色容量瓶中,用体积分数60%乙醇溶液定容,摇匀,按“1.3.2”节中色谱条件测定,并计算绿原酸总含量,结果见表5。由表5可知,绿原酸平均回收率为99.76%,RSD值为1.17%,<5%,表明该方法准确度高,符合实验对于定量的要求。
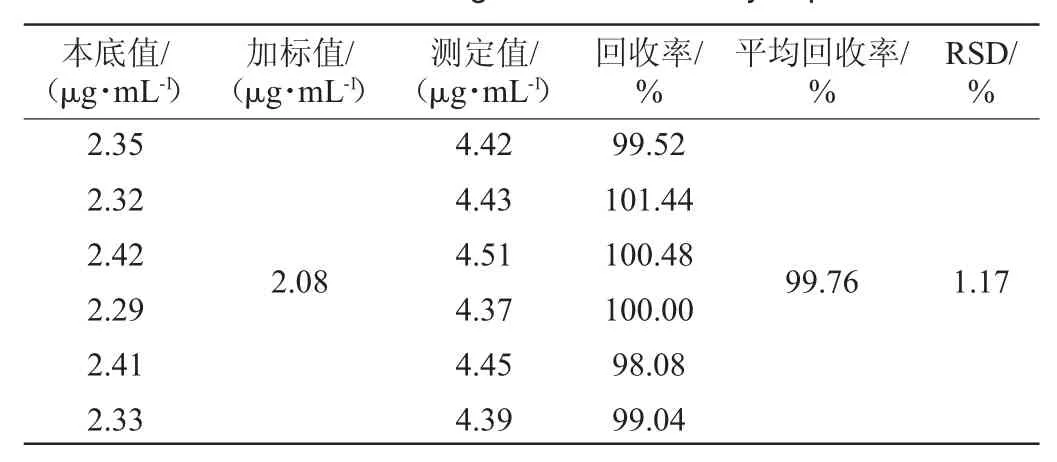
表5 加样回收率试验结果Table 5 Results of adding standard recovery experiments
2.7 样品中绿原酸含量的测定
取不同地区刺三加叶3批样品分别按“1.3.1”项下方法制备供试品溶液,再按照“1.3.2”节中色谱条件进样测定并计算绿原酸含量及相对标准偏差RSD(n=3),结果见表6。由表6可知,贵州渡马乡刺三加叶中绿原酸的含量最高为8.51 mg/g,广东恩平刺三加叶中绿原酸的含量最低为7.84 mg/g,可见不同地区刺三加叶中绿原酸的含量有一定的差异,但差异不明显。
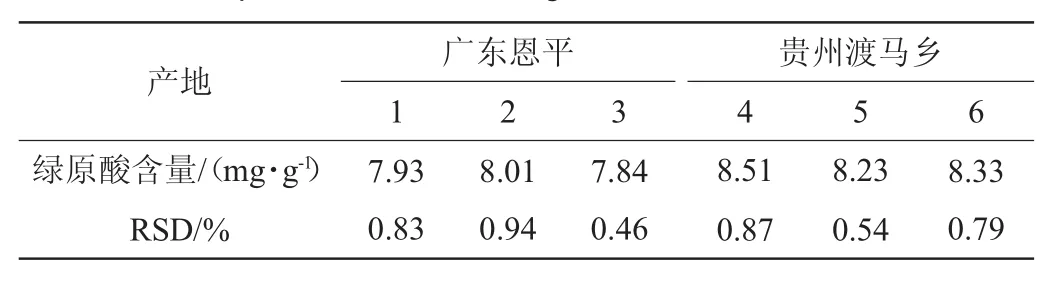
表6 不同地区样品中绿原酸含量的测定结果Table 6 Determination results of chlorogenic acid contents in the samples from different regions
3 结论
本研究确定了刺三加叶中绿原酸含量测定的最佳HPLC检测条件为ZORBAX SB-C18(4.6 mm×250 mm,5 μm)色谱柱;流动相选用甲醇-0.2%磷酸水溶液(15∶85,V/V);检测波长327 nm;流速1.0 mL/min;进样10 μL;柱温30℃。并在上述条件下进行了方法学验证,结果表明其方法操作简便,具有良好的精密度、准确度和稳定性。并在此基础上,测定了不同地区刺三加叶中绿原酸的含量,测定结果表明贵州渡马乡刺三加叶中绿原酸的含量最高为8.51 mg/g,不同地区刺三加叶中绿原酸的含量有一定差异,但差异不明显。
[1]李文芳,晏 丽,向 宁.刺三加叶组成成分与质量分析[J].天然产物研究与开发,2013,25(8):1077-1080.
[2]吴卫华,康 桢,欧阳冬生,等.绿原酸的药理学研究进展[J].天然产物研究与开发,2006,18(4):691-694.
[3]林春华,林伟君,谭 雪,等.野生蔬菜簕菜品质及急性毒性试验研究[J].现代食品科技,2009,25(2):201-202.
[4]向昌国,李文芳,聂 琴,等.甘薯茎叶中绿原酸提取方法的研究及含量测定[J].食品科学,2007,28(1):126-130.
[5]王 辉,田呈瑞,马守磊.绿原酸的研究进展[J].食品工业科技,2009,30(5):341-345.
[6]唐 柯,马 磊,徐 岩,等.超高效液相色谱法快速测定葡萄酒中酚酸[J].中国酿造,2015,34(12):157-161.
[7]张雪梅,谢金芮,陈玉甫,等.全酶解法提取杜仲叶中绿原酸及其含量测定[J].中国酿造,2016,35(10):149-152.
[8]ZHAO Q,WU X X,ZHOU J,et al.RP-HPLC determination of chlorogenic acid inLycium barbarumL.extract and its protective effect on retinal ganglion[J].Biomed Res India,2017,28(11):4869-4873.
[9]ZHANG Z H,PAN T W.HPLC determination of chlorogenic acid in Verbena officinalisL.extract and itsin vitroantibacterial activity[J].Biomed Res India,2017,28(9):3996-4001.
[10]LUO X L,LI N,XU M,et al.HPLC simultaneous determination of arbutin,chlorogenic acid and 6'-O-caffeoylarbutin in different parts of Vaccinium dunalianumwight[J].Biomed Res India,2015,29(20):1963-1965.
[11]SINGH A,BAJPAI V,KUMAR S,et al.Quantitative determination of isoquinoline alkaloids and chlorogenic acid inBerberisspecies using ultra high performance liquid chromatography with hybrid triple quadrupole linear ion trap mass spectrometry[J].J Sep Sci,2015,38(12):2007-2013.
[12]LI Z B,HUANG D N,TANG Z X,et al.Fast determination of chlorogenic acid in tobacco residues using microwave-assisted extraction and capillary zone electrophoresis technique[J].Talanta,2010,82(4):1181-1185.
[13]瞿万云,吴 艳,胡卫兵.基于纳米Nb2O5/石墨烯复合材料的增强效应电化学测定绿原酸[J].分析测试学报,2017,36(3):319-324.
[14]DAVID I.G,POPA D E,BULEANDRA M,et al.Cheap pencil graphite electrodes for rapid voltammetric determination of chlorogenic acid in dietary supplements[J].Anal Method,2016,8(35):6537-6544.
[15]蔡凌云,肖 娟,韩素菊,等.白簕叶中黄酮成分的鉴定和含量测定[J].绵阳师范学院学报,2010,29(11):78-84.
[16]蔡凌云,黎云祥,陈 焦,等.白簕根皮总黄酮提取工艺研究[J].食品科学,2009,30(4):15-18.
[17]钱 程,陈玉娥,黄 亮,等.HPLC法测定拳参中绿原酸和没食子酸含量[J].日用化学品科学,2016,39(7):25-28.
[18]张海娟,黄增荣,隆小华,等.HPLC法测定不同品种不同地区菊芋叶片中绿原酸的含量[J].天然产物研究与开发,2011,23(6):1107-1109.
[19]康 琛,李曼玲,殷文光,等.吴茱萸药材中绿原酸的含量测定[J].中国中药杂志,2008,33(17):2195-2196.
[20]史玉红.栀子药材中栀子苷和绿原酸含量测定方法研究[J].辽宁中医药大学学报,2013,15(6):38-40.
[21]李 娜,高晓燕,范 强,等.金银花提取液中绿原酸的快速定量[J].中国中药杂志,2007,32(4):312-314.
