SO32-活化S2O82-降解偶氮染料废水的机制研究
2018-03-24张昊楠段升飞汪婷婷
唐 海,张昊楠,段升飞,汪婷婷,李 洋
SO32-活化S2O82-降解偶氮染料废水的机制研究
唐 海*,张昊楠,段升飞,汪婷婷,李 洋
(安徽工程大学生物与化学工程学院,安徽 芜湖 241000)
为深度处理偶氮染料废水,以甲基橙(MO)为目标污染物,研究了亚硫酸盐活化过硫酸盐产活性物种的新型高级氧化处理方法, 并对活化机制、氧化机理及动力学理论进行分析.通过对SO32-/ S2O82-,S2O82-,SO32-3种体系进行降解对比和ESR等技术表征对比,发现亚硫酸盐能显著活化过硫酸根产生硫酸根自由基,其能氧化破坏MO偶氮双键形成的共轭体系,有较好的脱色降解效果.考察了亚硫酸盐和过硫酸盐摩尔比、过硫酸盐投加量、初始pH值对降解效果的影响, 结果表明当初始pH值为3.0,摩尔比1:1,投加量为20.0mmol/L、反应时间在300min下对MO降解率能达到96.1%,进一步发现该体系对初始pH值的适应范围较广(3.0~ 11.0).基于Box-behnken设计的响应面模拟和方差分析得到了可达显著水平的二次响应曲面模型,影响因子对MO降解的贡献排序为:过硫酸盐投加量>初始pH值>摩尔比.初始MO浓度动力学分析发现不同初始浓度下对MO的降解过程遵循准二级反应动力学规律,反应动力学常数从1.8212×10-4~ 2.4649×10-4min-1.另一方面发现升高反应温度可以促进体系对MO的降解,根据不同温度下活化过程的反应速率常数的阿累尼乌斯准二级反应的活化能计算结果(Ea=44.9kJ/mol),发现其相比常规金属活化方式较低,因此该体系对有毒有害的工业有机废水处理有潜在的商业应用价值.
偶氮染料废水;亚硫酸盐;过硫酸盐;活化;硫酸根自由基
印染行业是工业废水的排放大户,其成分复杂、水质变化大.近年来随着染料工业的发展和后整技术的进步,各种新型助剂、染料的大量使用,导致该类废水成分愈加复杂,特别是联苯胺型偶氮染料的使用,导致外排水中含有大量芳烃及杂环化合物为母体,并带有显色基团以及极性基团的有机组分,采用传统的物化、生化的工艺路线难以达标排放,属于典型的难降解工业废水[1-2].为实现国家节能减排要求,2015年实施新环保法之后,对印染废水的排放标准提出了更高的要求.甲基橙(MO,C14H14N3SO3Na)是染料废水中具有一定的代表性水溶性偶氮染料,发色基团为-N=N-(偶氮双键),不易被一些常规的方法所降解,所以更易引起严重的环境污染问题.
当前国内外对含有偶氮染料废水处理进行了大量研究,在高级氧化法方面.Punzi[1]用photo-Fenton技术处理东西雷马素红染料厌氧生物膜出水,发现Fe2+和H2O2最低投加量分别在1mM和10mM条件下,氧化处理出水COD低于18 mg/L,去除率高于90%;刘双柳等[2]用催化还原技术将Cu/Cu2O/C作为催化剂用于亚甲基蓝等偶氮染料的的降解,重复利用5次,降解率仍在99%以上,表现出了良好的催化活性和稳定性.特别是偶氮染料废水的降解和矿化研究日益受到重视,探索低成本、高效染料废水处理技术并付诸实践势在必行.
基于过硫酸盐活化产生强氧化性硫酸根自由基(SO4•-)的高级氧化技术受到较多关注,己被广泛用于修复受到污染的地下水和土壤[3].由于SO4•-的氧化能力强(0= +2.5V ~ +3.1V/NHE),超过了羟基自由基(×OH),对水中有机物具有无选择性和持续性的氧化,理论上可降解大部分有机污染物,具有较快的反应速率,而且其氧化产物SO42-不影响后续生物处理.特别是过硫酸盐易储存,环境友好,安全稳定易于控制,价格低有很好的商业前景,因此近年来基于硫酸根自由基高级氧化技术成为处理有毒有害废水新途径[4-8].当前过硫酸盐活化技术发展非常迅速,除了常见热活化、过渡金属离子活化、UV活化、碱活化、超声波活化等[3,5,7,9],又不断涌现出一些新型的活化技术,特别以铁氧化物材料[10-11](CuO-Fe3O4、碳酸铁)、零价金属[12](Fe0、Zn0)、碳基材料[13-14](活性炭、碳纳米管)等以及包括以上述材料为基础改性合成的新型材料等成为国内外的研究热点.
本研究提出了一种新型的过硫酸盐的活化方法,用亚硫酸盐活化过硫酸盐(PS)产生硫酸根自由基(SO4•-).通过ESR技术表明SO32-能显著活化S2O82-,以甲基橙作为目标污染物,结合紫外光谱和红外光谱详细分析其活化机制和降解机理.用序批式试验考察了SO32-和S2O82-摩尔比、体系初始pH值、过硫酸盐投加量、MO初始浓度以及反应温度对MO去除效果的影响.结合反应动力学拟合结果获得其活化能,使用响应面方法对反应条件进行了优化研究,获得主要影响因素.本研究不仅可推动过硫酸盐活化体系的发展,而且为染料废水的深度处理提供可行的解决方案和可靠的技术路线.
1 材料与方法
1.1 仪器试剂与分析方法
实验中所有试剂均为分析纯.所用仪器有数显恒温磁力搅拌器(85-2B,华伟仪器)和pH 计 (PHS-3C,上海雷磁)等.自由基鉴定采用ESR波谱仪(JES-FA200,日本电子).反应前后水样MO降解率及表征采用紫外分光光度仪(T6,北京谱析通用)测定.红外表征采用傅立叶变换红外光谱仪(IRPrestige-21,日本岛津),其分析条件为:取反应后离心的上清液20mL放入小烧杯中,再取0.2g KBr晶体溶解于其中,然后将烧杯置于真空干燥24h,使反应体系中的有机分子吸附于KBr分子表面,样品干燥后压片再进行红外分析.采用Design Expert V8.0进行响应面优化分析,以摩尔比(SO32-)/(S2O82-)、初始pH值和S2O82-投加量为自变量,MO降解率为响应,根据Box- Behnken中心组合原理,设计实验.
1.2 试验步骤
采用MO配制一定浓度的250mL模拟偶氮染料废水,设定温度和搅拌数值,用0.1mol/L NaOH和H2SO4调节反应体系初始pH至设定值,确定并加入所需Na2SO3和Na2S2O8,在一定时间下进行活化及氧化过程,每隔一段时间取样,迅速用0.45μm滤膜过滤后,收集滤液在紫外下测定其吸光度(MO最大吸收波长473nm处),从而确定MO浓度.去除率MO通过公式(1)计算得到.
MO=(0-e)/e×100% (1)
式中:MO为MO的降解率;0(mg/L)和e(mg/L)分别为MO的初始浓度和反应结束时浓度.
2 结果与讨论
2.1 降解对比研究及活化机理分析
实验对比了SO32-活化S2O82-、单一S2O82-和单一SO32-三种体系对MO的降解.MO初始浓度0=10mg/L,(SO32-)和(S2O82-)投加量均为10mM,初始pH值为3.0,结果如图1所示.由图可知,同一实验条件下,从MO降解率来看,SO32-对S2O82-有明显的活化效果.在前30min内,SO32-/ S2O82-活化体系的MO降解率比单一S2O82-要高35.26 %,在60min内降解率接近88.55 %;而单一S2O82-的降解效率在60min才达到67.52,两者MO降解率相差近21.03 %.而单一SO32-体系MO降解率很低,60min低于5.93 %.
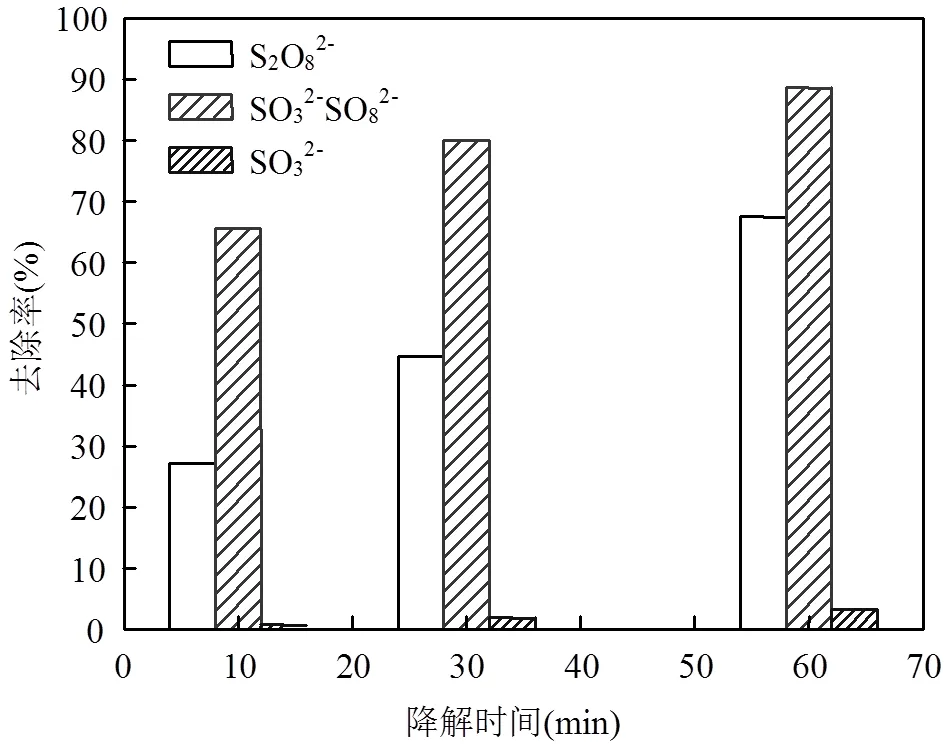
图1 3种体系对MO降解率对比
为进一步证实SO32-/S2O82-活化机制,用测定短寿命自由基的电子自旋共振(ESR)仪予以鉴定.将自旋捕捉剂DMPO加入到三种体系中,生成寿命较长的自旋加合物(DMPO +R×→DMPO-×R),图2为三种体系的ESR波谱(反应时间为5min).从图中可以看出,在SO32-/S2O82-活化体系下,ESR谱图显示有较强的DMPO-×OH(1:2:2:1的四重峰)和DMPO- SO4•-加合物的ESR波谱的特征信号峰[15];由于在水溶液中SO4•-能迅速与水反应转化成×OH,而且×OH的自旋要高于SO4•-,所以×OH信号相对硫酸根自由基强.而单一SO32-体系中,ESR谱图没有信号出现;单一S2O82-体系有较弱的DMPO-×OH信号.由此说明了SO32-能显著活化S2O82-产生SO4•-,同时·OH自由基为该体系活化反应的主要氧化中间体.
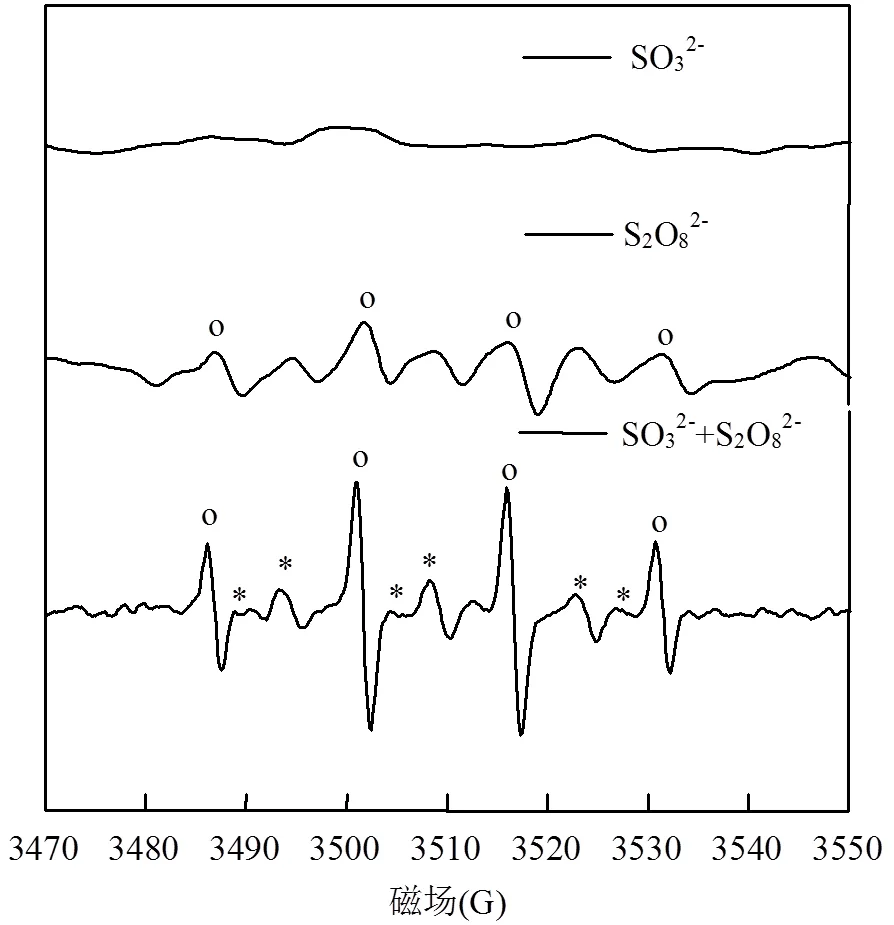
图2 DMPO捕获硫酸根自由基ESR谱图
反应5min;o,DMPO- •OH;*,DMPO- SO4•-
进一步分析MO降解后分子结构变化,对MO原水、SO32-/S2O82-和单一S2O82-进行UV和FT-IR扫描光谱分析,结果分别见图3和图4.可以看出,原MO在473nm附近出现MO分子偶氮结构π-π*跃迁引起的强吸收峰[16],在270nm和312nm处是因苯环共轭体系π-π*引起2个较弱的吸收峰;在单一S2O82-氧化体系下,MO在473nm处的峰高明显降低,在270nm处的吸收峰未明显下降;而在SO32-/S2O82-体系作用下,MO在整个氧化过程中473nm、270nm和310nm处的吸收峰几乎完全消失,而且偶氮键吸收峰明显要比苯环共轭体系下降的快.同样从红外结果来看,原水MO在1460cm-1附近是偶氮键的伸缩振动峰[17],1518cm-1附近为苯环骨架上的C=C振动吸收峰,1118cm-1位推测是N原子连接与苯环C-N特征吸收峰,1365cm-1是-SO3-的对称与非对称振动峰,而通过SO32-/S2O82-体系结果来看,1460cm-1处偶氮振动吸收与原水样相比几乎完全消失,1518cm-1处苯环的吸收峰和1365cm-1处特征峰明显强度降低,以上结论均证明了MO结构中的共轭发色体系已经破坏.
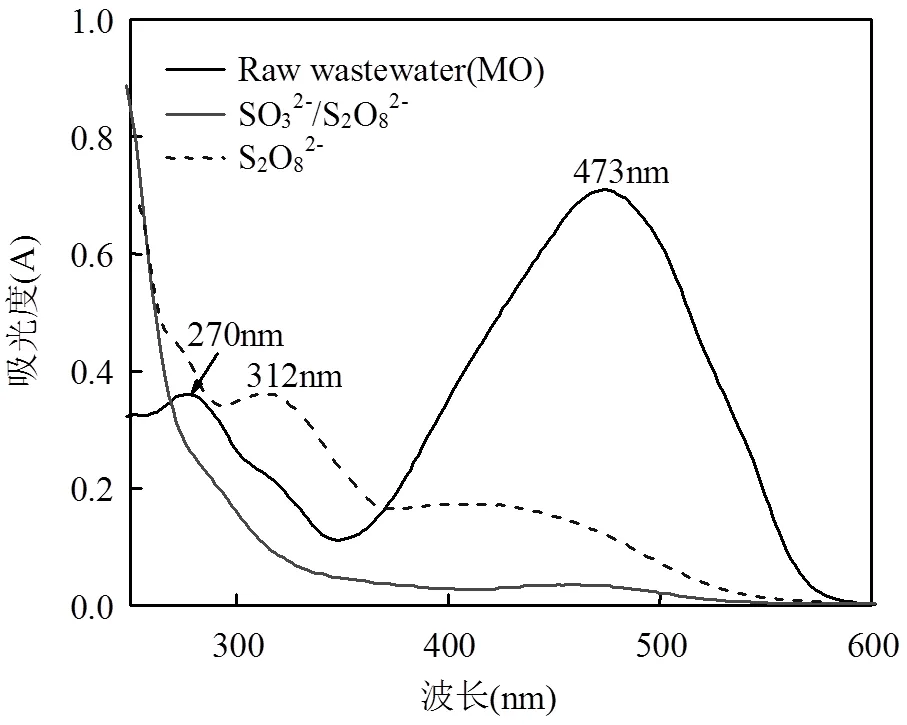
图3 紫外扫描光谱图对比
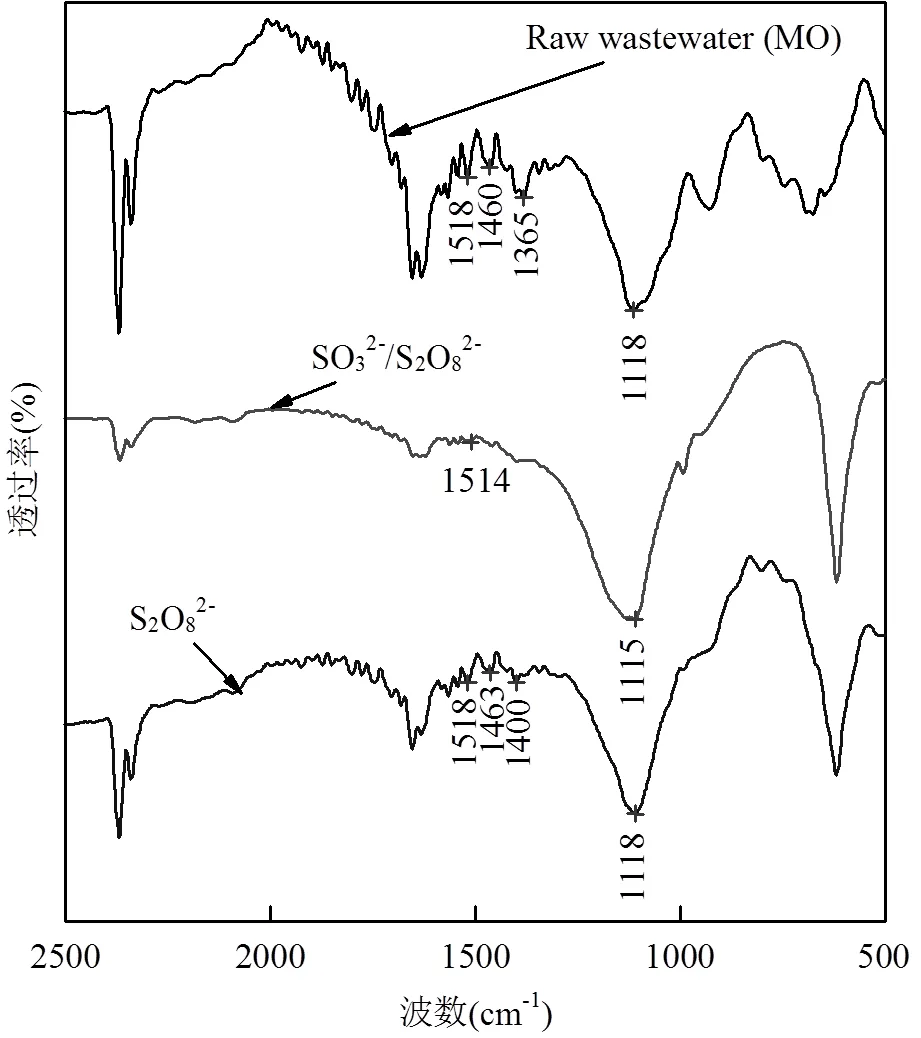
图4 红外光谱图对比
可以推测SO32-活化S2O82-过程中产生了更多的活性物种(强氧化性硫酸根自由基SO4•-),其活化机理可以用式(2)予以描述.
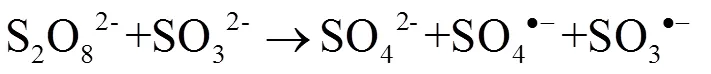
由于SO4•-具有很强的氧化活性及较高的电子亲和能,首先进击电子云密度较高的偶氮基团,其活泼的单电子诱导了偶氮上孤对电子向单电子转变,所形成的含四个单电子活泼的过渡态与4个质子迅速结合,从而形成了两个氨基,破坏了MO偶氮结构,导致MO被SO4-降解,转化为其他产物,溶液中MO浓度也随之降低.其初步降解反应机理见图5所示.
2.2 单因素影响实验
2.2.1 物质的量浓度比的影响 当反应体系MO初始浓度MO=60mg/L,初始pH=3.0,分别考察不同摩尔比(SO32-):(S2O82-)为5mmol/ L:5mmol/L、2.5mmol/L:5mmol/L和5mmol/L: 7.5mmol/L对MO降解率的影响,结果见图6.可以看出,反应300min内,当SO32-初始浓度从0.25mmol/L提高到0.5mmol/L时,MO降解率明显上升,从63.2 %增加到79.1 %.而固定(SO32-)为5mmol/L,随着S2O82-浓度进一步提高,MO降解率反而略有降低,从79.1%降低到76.7%.这表明适量的SO32-会促进S2O82-的活化,强化对MO的降解,当S2O82-过量时会对MO降解反而起到抑制作用.
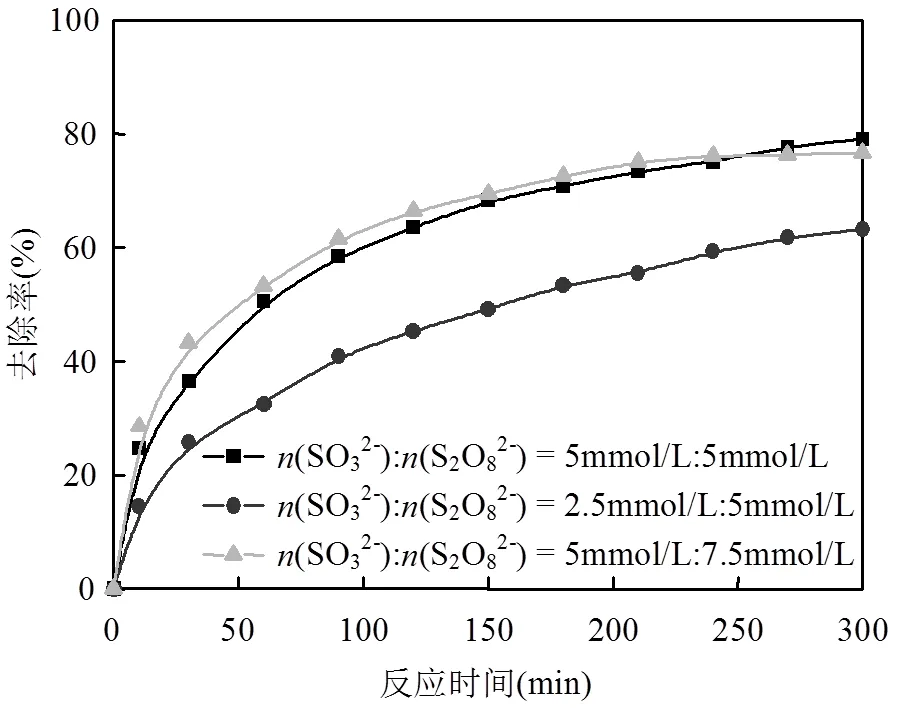
图6 摩尔比对MO降解率的影响
由于SO32-与S2O82-反应分两步进行,SO32-为电子供体,首先它会催化分解S2O82-,产生一定稳态浓度的SO4•-氧化降解MO.但是当S2O82-浓度较高时,产生的SO4•-同时会与溶液中存在的S2O82-发生淬灭,从而消耗了部分SO4•-[3],同时在这一过程中MO与SO32-对SO4•-产生了竞争作用,也抑制了MO的降解.因此,适量的SO32-对有效活化S2O82-是非常必要的,后续实验选择(SO32-):(S2O82-)=5mmol/L:5mmol/L进行.
2.2.2 初始pH值的影响 图7为(SO32-) =(S2O82-)= 5.0mmol/L,初始浓度MO=60mg/L条件下,不同pH值条件下 MO降解率的变化趋势.结果表明,当pH=2.0时,其降解率最低,反应300min只达到了65.54%;而当pH值范围在3.0~9.0时,随着初始pH值的增加,MO的去除率呈略微下降趋势,在pH值为3.0时降解去除率为87.63%,当pH值为4.0时为84.89%,较pH值为3.0时低.但pH值为6.0、9.0时甲基橙的降解去除率也分别达到了84.06%和81.71%.而pH值为11.0时,甲基橙降解去除率为84.82%,与pH值为4.0时相当,也说明了SO32-/S2O82-活化体系对初始pH值的适应范围较广.

图7 初始 pH对MO降解率的影响
这种现象主要与MO形态有关,在酸性条件下,MO中的氮原子质子化后,形成了醌式结构,键能降低,更易被催化氧化[18],所以酸性条件下更有利于MO的降解.此外,在酸性条件下SO4•-能够按照式(3)和式(4)被酸激发,而pH值较高的情况下主要是SO4•-与H2O或者OH-反应生成氧化活性较低的×OH[11](式5),所以MO随pH值增加去除率呈下降趋势.考虑S2O82-初始体系为酸性,选取pH=3.0为宜.
SO4•-+ H2O → HSO4–+ •OH (3)
S2O82-+ H+→ HS2O8—→SO4•-+ SO42-+ H+(4)
SO4•-+ OH-→ SO42-+ •OH (5)
与此同时,从pH值的变化趋势来看(见图8),只有pH值在2.0的情况下整个反应体系过程中的pH值几乎没有变化,其他初始pH值为在整个反应时间内具有较大的变化,初始pH值分别为11.0、9.0、6.0、3.0、2.0时经过300min反应后分别降到了6.53、4.06、2.63、2.48、1.86.这种现象可能是质子的释放与酸性中间产物的形成[19].
2.2.3 过硫酸盐投加量的影响 图9为初始pH=3.0,MO初始浓度MO=60mg/L,固定(SO32-)/(S2O82-)=1.0,不同S2O82-浓度下对MO降解过程的影响.从图9可以看出,在考察的S2O82-浓度范围内,当S2O82-浓度为5.0mmol/L时,反应300min后,MO降解率从0%达到75.2%.随着过S2O82-初始浓度从5.0mmol/L增加至20.0mmol/L,MO降解率也相应增加,从0达到96.1%,这也说明了随着S2O82-的增加,氧化性的活性物种得到了相应的增加, MO去除率的变化趋势与其他金属离子例如Fe2+活化方式的结果类似[8].而当S2O82-浓度继续增加到25mmol/L时,MO降解率为95.6%,略有下降,趋于平稳.
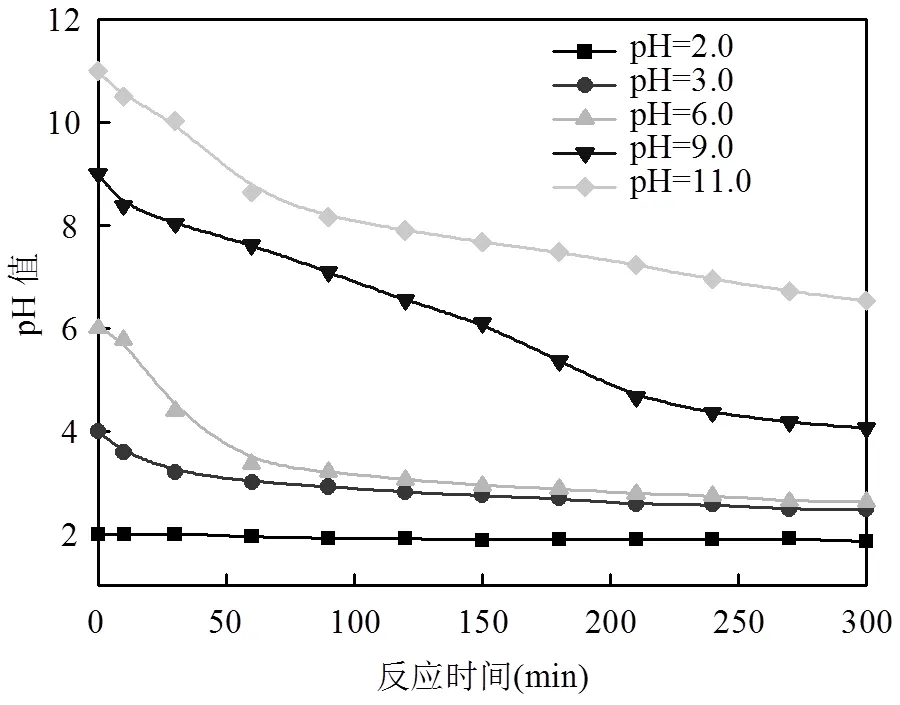
图8 不同初始pH值在反应过程中的变化

图9 过硫酸盐投加量对 MO降解率的影响
2.3 响应面优化
根据单因素结果,确定Box-Behnken模型较优化水平为:(SO32-):(S2O82-)为0.5~0.75,(S2O82-)为5.0~25.0mmol/L,初始pH值为2.0~ 11.0,设定反应时间为60min,试验设计和MO降解率见表1,回归方程的方差分析结果见表2.根据BBD统计学要求,从不同模型方差分析中的均方及检验结果综合来看,整体模型为显著(<0.05),多元相关系数2=0.8540,说明回归方程与试验拟合较好,模型的可信度和精密度较高.进一步分析可见,单因素中过硫酸盐投加量对MO的去除有极显著的影响(<0.01).影响因子对MO降解的贡献排序为:过硫酸盐投加量>初始pH值>摩尔比.同时基于Box-behnken设计的响应面模拟和方差分析得到了二次响应曲面模型,结果如图10所示.对于三维响应曲面图,在图中颜色越深表明效果越显著,去除效果越好[20].由图10可知,pH值越高或S2O82-浓度越高,MO降解率越高,尤其是pH值在10.0~11.0时,过硫酸钠浓度在20~25mmol/L时,曲面颜色最深,MO去除效果最好.
表1 Box-behnken试验设计与结果

表2 回归方差分析
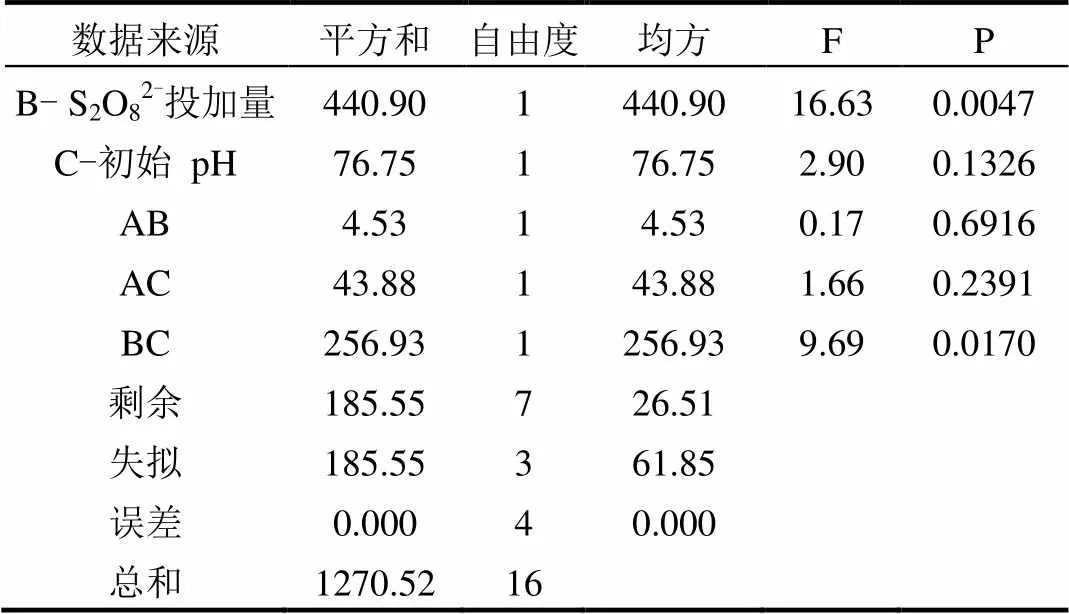
续表2

图10 多因子对MO降解效果影响响应曲面
2.4 初始浓度影响及反应动力学计算
图11为初始pH=3.0,(SO32-)=(S2O82-)= 5.0mmol/L下不同初始MO浓度(10~60mg/L)对降解过程的影响.由图可知,随着MO浓度的升高,其降解率呈下降趋势.由图11数据作图,对不同初始浓度下的数据进行拟合,相应的反应动力学拟合,从结果可以看出SO32-/S2O82-活化体系对MO降解的化学动力学均更加符合伪二级动力学方程,形式如式(6)所示,结果见图12:
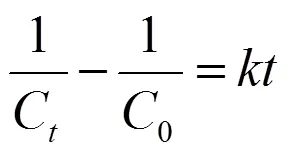
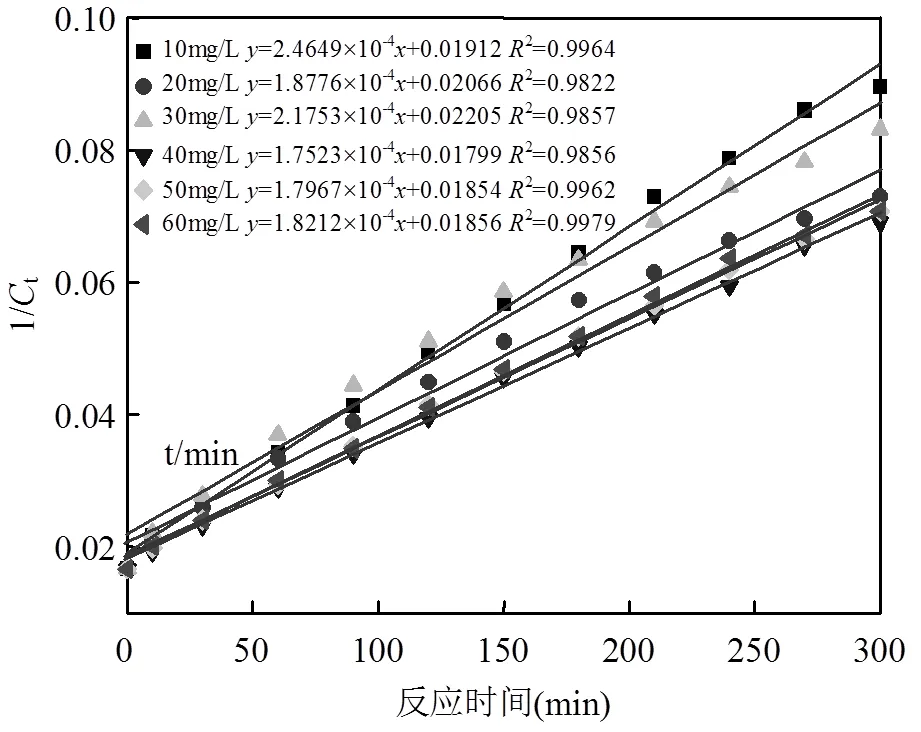
图12 伪二级反应动力学拟合结果
所得的参数于图12列出,线性相关系数2=0.9822~0.9979,其中代表速率常数.当浓度从10mg/L上升至60mg/L时,可知其反应速率常数及降解去除率应该呈梯度递减,从2.4649´10-4min-1降低至1.8212´10-4min-1.这是由于在反应过程中,MO浓度的增加,导致污染物之间产生竞争,降解不彻底将增加中间产物种类,这样也消耗体系中产生的SO4•-,造成反应速率的降低.
2.5 温度影响及反应活化能计算
当温度由298K增加至318K,初始pH=3.0,(SO32-)=(S2O82-)=5.0mmol/L,MO初始浓度MO=60mg/L,考察温度对MO降解效率的影响规律.从图13中看出,随着温度升高,氧化体系的反应速率增大,MO的去除效率也得到相应提高.随着的增大,值得到了提高,当=298K时,= 0.00026min-1,当增大到318K时,= 0.00072min-1,相应的MO降解率提高了19.13%,体系的氧化性能显著增强.根据不同反应温度下速率常数的变化规律,结合Arrhenius公式(式7),可以分析求得实验条件下的反应活化能E(kJ/mol)和频率因子(min-1).
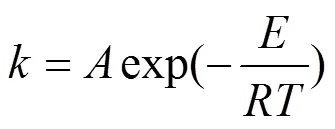
由图14看出,ln-1/呈良好的线性关系,对得到的多元回归方程式中相关系数换算后,得到实验条件下以MO表示有机物的伪二级反应的反应活化能E为44.9kJ/mol.据报道常规热活化断裂过硫酸盐的O-O需要的活化能为140.2kJ/ mol,Fe(II)催化需要12.5kca/mol[21],紫外活化过硫酸盐降解双酚A的活化能为26kJ/mol[22],活性炭催化过硫酸钠降解金橙G的活化能为22.61kJ/mol[13],尽管目标污染物结果的活化能有所差异,但是相比上述,用SO32-/S2O82-体系降解MO相对较低,而且很容易实现.
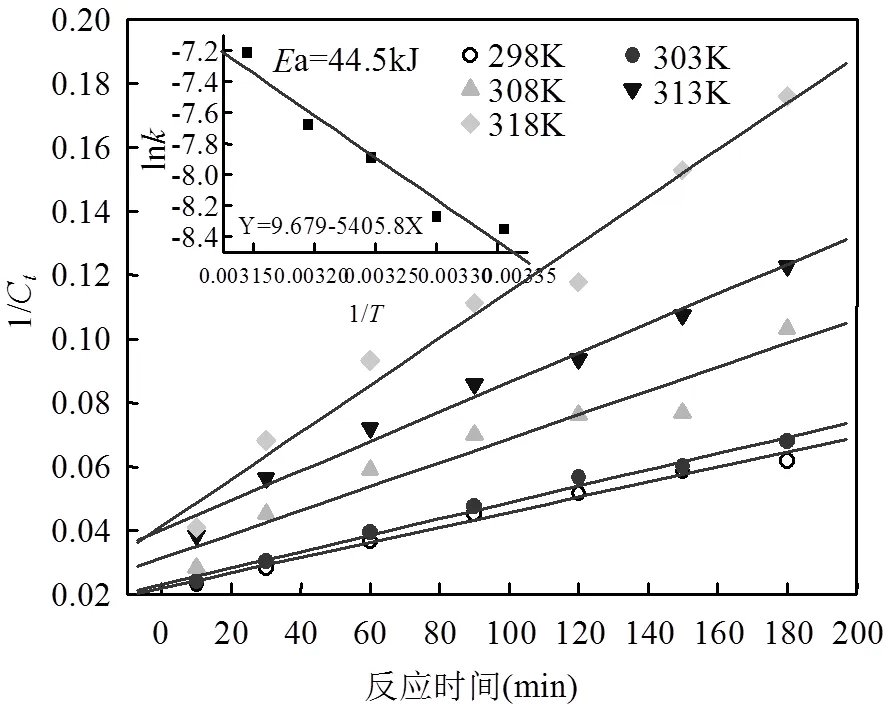
图14 活化能拟合结果
3 结论
3.1 亚硫酸盐能显著活化过硫酸盐产硫酸根自由基,诱导偶氮上孤对电子向单电子转变,所形成活泼的过渡态迅速与质子结合,破坏MO偶氮双键形成的共轭体系,有较好的降解效果.在MO初始浓度60mg/L,当初始pH值为3.0,亚硫酸盐和过硫酸盐摩尔比1:1,投加量均为20mmol/L、反应时间在300min下对MO降解率能达到96.1%.该体系对初始pH值的适应范围较广(3.0~11.0),升高反应温度可以促进体系对MO的降解.
3.2 基于Box-behnken设计的响应面模拟和方差分析得到了可达显著水平的二次响应曲面模型,影响因子对MO降解的贡献排序为:过硫酸钠浓度>初始pH值>摩尔比.发现不同初始浓度下对MO的降解过程遵循准二级反应动力学规律,反应动力学常数为0.00026~0.00072min-1.反应动力学常数为1.8212´10-4~2.4649´10-4min-1.
3.3 根据不同温度下的反应速率常数下活化过程的阿累尼乌斯准二级反应的活化能的计算结果(E=44.9kJ/mol),发现其相比常规金属活化方式较低,该体系对有毒有害的工业有机废水有潜在的商业应用价值.
[1] Punzi M, Anbalagan A, Börner R A, et al. Degradation of a textile azo dye using biological treatment followed by photo- Fenton oxidation: Evaluation of toxicity and microbial community structure [J]. Chemical Engineering Journal, 2015, 270(15):290-299.
[2] 刘双柳,施春红,牛红云,等.纳米铜复合材料催化还原染料废水的研究[J]. 中国环境科学, 2015,35(3):764-769.
[3] Matzek L W, Carter K E. Activated persulfate for organic chemical degradation: A review [J]. Chemosphere, 2016,151: 178-188.
[4] 何洋洋,唐素琴,康婷婷,等.响应面法优化硫酸根自由基高级氧化深度处理渗滤液生化尾水[J]. 中国环境科学, 2015,35(6): 1749-1755.
[5] Zhang B T, Zhang Y, Teng Y, et al. Sulfate radical and its application in decontamination technologies [J]. Critical Reviews in Environmental Science & Technology, 2015,45(16): 1756-1800.
[6] 唐 海,徐建平,安 东,等.TiO2/ZSM-5m光催化耦合过硫酸盐降解焦化尾水的研究[J]. 中国环境科学, 2015,35(11):3325- 3332.
[7] 朱思瑞,高乃云,鲁 仙,等.热激活过硫酸盐氧化降解水中双酚A [J]. 中国环境科学, 2017,37(1):188-194.
[8] Zhu L, Ai Z, Ho W, et al.Core–shell Fe-Fe2O3nanostructures as effective persulfate activator for degradation of methyl orange [J]. Separation & Purification Technology, 2013,108(16):159-165.
[9] Xie P, Ma J, Liu W, et al.Removal of 2-MIB and geosmin using UV/persulfate: contributions of hydroxyl and sulfate radicals [J]. Water Research., 2015,69:223.
[10] Bennedsen L R, Muff J, Søgaard E G. Influence of chloride and carbonates on the reactivity of activated persulfate [J].Chemosphere,2012,86(11):1092-1097.
[11] Lei Y, Chen C S, Tu Y J, et al. Heterogeneous degradation of organic pollutants by persulfate activated by CuO-Fe3O4: mechanism, stability, and effects of pH and bicarbonate ions [J]. Environmental Science & Technology, 2015,49(11):6838-6845.
[12] Li H, Guo J, Yang L, et al. Degradation of methyl orange by sodium persulfate activated with zero-valent zinc [J]. Separation & Purification Technology. 2014,132:168-173.
[13] 陈家斌,魏成耀,房 聪,等.碳纳米管活化过二硫酸盐降解偶氮染料酸性橙7 [J]. 中国环境科学, 2016,36(12):3618-3624.
[14] 王 莹,魏成耀,黄天寅,等.氮掺杂碳纳米管活化过一硫酸盐降解酸性橙AO7 [J]. 中国环境科学, 2017,37(7):2583-2590.
[15] Zhang G, Wu Z, Liu H, et al. Photoactuation healing of α-FeOOH@g-C3N4 catalyst for efficient and stable activation of Persulfate. [J]. Small, 2017.
[16] 孔令国,王 玲,薛建军.负载型三维粒子电极降解甲基橙模拟废水研究[J]. 中国环境科学, 2010,30(4):516-521.
[17] Wu R, Qu J. Removal of water-soluble azo dye by the magnetic material MnFe2O4[J]. Journal of Chemical Technology & Biotechnology Biotechnology, 2005,80(1):20-27.
[18] 占昌朝,曹小华,严 平,等.微波促进Fe2+-K2S2O8体系快速降解甲基橙废水的研究[J]. 水处理技术, 2013,39(8):43-46.
[19] Qi C, Liu X, Ma J, et al. Activation of peroxymonosulfate by base: Implications for the degradation of organic pollutants [J]. Chemosphere. 2016,151:280-288.
[20] 侯韦竹,丁 晶,赵庆良,等.响应面法优化电氧化-絮凝耦合工艺深度处理垃圾渗滤液[J]. 中国环境科学, 2017,37(3):948-955.
[21] Kolthoff I M, Miller I K. The chemistry of persulfate. I. The kinetics and mechanism of the decomposition of the persulfate ion in aqueous medium1 [J]. Journal of the American Chemical Society, 1951,73(7):1-30.
[22] Huang Y F, Huang Y H. Identification of produced powerful radicals involved in the mineralization of bisphenol A using a novel UV-Na2S2O8/H2O2-Fe(II,III) two-stage oxidation process.[J]. Journal of Hazardous Materials. 2009,162(2/3):1211.
Mechanism research for degradation of azo dying wastewater based on persulfate activated by sulphite.
TANG Hai*, ZHANG Hao-nan, DUAN Sheng-fei, WANG Ting-ting, LI Yang
(School of Biochemical Engineering, Anhui Polytechnic University, Wuhu 241000, China)., 2018,38(3):959~967
For the advanced treatment of azo dyeing wastewater, a novel advanced oxidation technology was studied for the the production of active species based on persulfate activated by sulfite using methyl orange (MO) as the target pollutant, and its activation mechanism, oxidation theory and kinetic were analyzed. Through the degradation comparison of the SO32-/S2O82-, S2O82-, SO32-three systems and ESR and other technical characterization, it is revealed that persulfate can be activated by sulfite significantly producing sulfate radicals to destroy the conjugated system of MO azo double bond, which has a better decolorization and degradation effects. The effects of the molar ratio of sulfite to persulfate, dosage of persulfate, initial pH on the degradation of MO were investigated. The results showed that the percent removal rate of MO reached 96.1% under the conditions: the initial pH of 3.0, the molar ratio of sulfite to persulfate of 1:1, the dosage of persulfate of 20mmol/L at the reaction time of 300min. It is further found that the system has a wide range of adaptation to the initial pH (3.0~11.0). The response surface based on Box-behnken model was simulated and the analysis of variance was two times the response surface model up to significant level, contributing factor on the degradation of MO as follows: initial dosage of persulfate > pH value >molar ratio. The oxidation process analyzed by kinetics under the different initial concentrations was found that it fits well perso second-order reaction kinetics. The reaction kinetic constants vary from 1.8212×10-4~ 2.4649×10-4min-1. On the other hand, the increasing of temperature can promote the removal of MO, according to the perso second-order reaction rate constants at different temperatures were calculated Arrhenius activation energy Eaoxidation process (44.9kJ/mol), its value is relatively low compared with the conventional metal activation method, which reveals it has a potential commercial value in the treatment of toxic and harmful industrial organic wastewater.
azo dye wastewater;sulfite;persulfate;activation;sulfate radical
X703.5
A
1000-6923(2018)03-0959-09
唐 海(1976-),男,安徽安庆人,教授,硕士,主要从事水处理技术研究.发表论文30余篇.
2017-07-24
国家自然科学基金资助项目(51274001);安徽省自然科学基金资助项目(1608085ME118);安徽省优秀人才基金资助项(gxyqZD2016120)
* 责任作者, 教授, newth76@qq.com
