GC-MS/SIM法测定盐酸鲁拉西酮片中甲磺酸酯类物质残留量
2018-03-23,2,2*
,2 ,2*
(1.北京市理化分析测试中心,有机材料检测技术与质量评价北京市重点实验室,北京100089;2.北京市科学技术研究院分析测试技术重点实验室,北京 100089)
甲磺酸酯类化合物主要来自于合成过程中的反应副产物,其作为一类基因毒性杂质对DNA具有潜在的破坏性,一直备受关注[1]。药物生产过程中,醇类物质一般作为溶剂参与药物合成,甲磺酸常作为反离子试剂与药物活性成分形成稳定的共轭盐类物质,以改善其溶解性、吸收性等理化特性。在药物合成过程中,甲磺酸会与醇类物质发生副反应,生成具有基因毒性的甲磺酸酯类物质,此类物质难以完全从合成体系中除去,具有很强的致畸、致癌作用[2,3]。研究表明,甲磺酸酯类物质可作用于DNA分子的嘌呤基团,生成烷基化嘌呤,破坏DNA双链的空间构象和稳定,诱发机体基因突变和癌症[4,5]。
药物中基因毒性杂质的残留问题,已成为制药行业所面临的重大挑战,自2007年甲磺酸奈非那韦事件后,美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA),均相继规定基因毒性杂质的每日摄入量不能超过1.5μg[6-7]。盐酸鲁拉西酮(lurasidone hydrochloride)是一种新型治疗成人精神分裂症的药物,该药物中残留的甲磺酸酯类物质会对患者的身体健康造成损害,因此,建立一个高灵敏度、操作简便、可行性强的分析方法,来检测此类物质,对保证药物安全至关重要。近年来,国内外虽已有较多文献报道甲磺酸酯类物质的检测方法,例如GC、GC-MS、HPLC-MS、HPLC-MS/MS等方法[8-13],但关于使用GC-MS同时检测盐酸鲁拉西酮片中甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的方法,目前未见报道。本研究建立了GC-MS/SIM直接进样检测盐酸鲁拉西酮片中甲磺酸酯类物质的方法,该方法不需要对样品进行繁琐的前处理操作,能同时检测甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的残留量,在保障药物安全方面有较重要的应用价值。
1 实验部分
1.1 仪器与试剂
GC-MS QP2010 Ultra气相色谱-质谱联用仪,日本Shimadzu公司;XPE105分析天平,瑞士Mettler Toledo公司;甲磺酸甲酯(纯度>98.0%)、甲磺酸乙酯(纯度>99.0%)、甲磺酸异丙酯(纯度98.0%),日本TCI公司;甲醇(色谱纯),美国Fisher公司;盐酸鲁拉西酮片,某制药公司提供。
1.2 标准溶液配制
分别准确称取适量甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯标准物质,甲醇溶解定容,配制得质量浓度为1000mg/L的单标准贮备液。再分别准确移取3种单标准贮备液各0.1mL,混合于100mL容量瓶中,甲醇稀释定容,配制成1mg/L的混合标准贮备液,使用时再用甲醇逐级稀释至所需浓度的混合标准溶液。所有标准溶液在4℃冰箱中贮存。
1.3 样品处理
按盐酸鲁拉西酮有效成分40mg的剂量计,取盐酸鲁拉西酮片适量,置于5mL离心管中,加入甲醇2.0mL,涡旋振荡至药片完全溶解,0.22μm有机膜过滤,即得样品制备液。
1.4 GC-MS分析条件
GC条件:DB-624毛细管色谱柱(30m×0.32 mm×1.8μm);进样口温度250℃;载气为高纯He(纯度>99.999%);分流进样,分流比2∶1;进样量1.0μL;柱流量3.0mL/min;升温程序,初始柱温120℃保持7min,20℃/min升至240℃后保持2min。
MS条件:电子轰击离子源(EI);电子能量70eV;离子源温度230℃;接口温度250℃;溶剂延迟时间1.5min;选择离子监测(SIM)模式;甲磺酸甲酯: 80m/z为定量离子,65m/z、109m/z为定性离子;甲磺酸乙酯:109m/z为定量离子,80m/z、65m/z为定性离子;甲磺酸异丙酯:123m/z为定量离子,124m/z为定性离子。
2 结果与讨论
2.1 提取溶剂选择
分析比较了甲醇、乙腈、二甲基甲酰胺和二甲基亚砜作为提取溶剂,对检测结果的影响。研究表明,二甲基甲酰胺和二甲基亚砜虽对盐酸鲁拉西酮片的溶解度较好,但甲磺酸酯类物质在这两种溶剂体系下响应值却较低,会降低检测灵敏度。甲醇与乙腈相比,两者对盐酸鲁拉西酮片的溶解能力相近,但在甲醇体系中甲磺酸酯类物质响应值更高,且峰型也较理想。综合考虑,本方法选择甲醇作为提取溶剂。
2.2 特征离子选择
综合考虑目标物的特征离子和药物基质的干扰离子,在保证药物其它成分不干扰检测的前提下,尽量选取丰度较高、质荷比较大的特征离子,进行GC-MS/SIM分析。为了尽可能提高检测灵敏度,减小目标物间的干扰,本研究在选择离子监测模式下,对三种目标物的特征离子,进行分段采集。为了有效降低基质效应,在定量离子选择方面,甲磺酸甲酯选择了质荷比较大、丰度最高的80m/z;甲磺酸乙酯未选择丰度最高的79m/z,而选择了质荷比较大、丰度次之的109m/z;甲磺酸异丙酯也舍弃了质荷比较小、丰度最高的43m/z,而选择了质荷比较大、丰度次之的123m/z进行定量分析。在定性离子选择方面,一般应选择两个辅助定性分析,但由于甲磺酸异丙酯的特征离子会引起较强的基质效应,干扰其分析,所以只选取了质荷比较大但丰度较低的124m/z作为定性离子;同样,为了有效降低基质效应,甲磺酸甲酯选择了65m/z、109m/z,甲磺酸乙酯选择了80m/z、65m/z进行定性分析。图1、图2分别为混合标准溶液和样品制备液的GC-MS/SIM选择离子流图,从图上可知,盐酸鲁拉西酮片中的其它成分不会对目标物的检测造成干扰,该药物中未能检测出目标物,本方法所选取的特征离子适宜,能有效消除该药物的基质效应,适用于该药物中甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的测定。
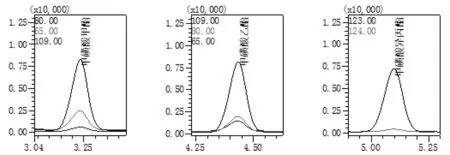
图1 混合标准溶液GC-MS/SIM选择离子流图
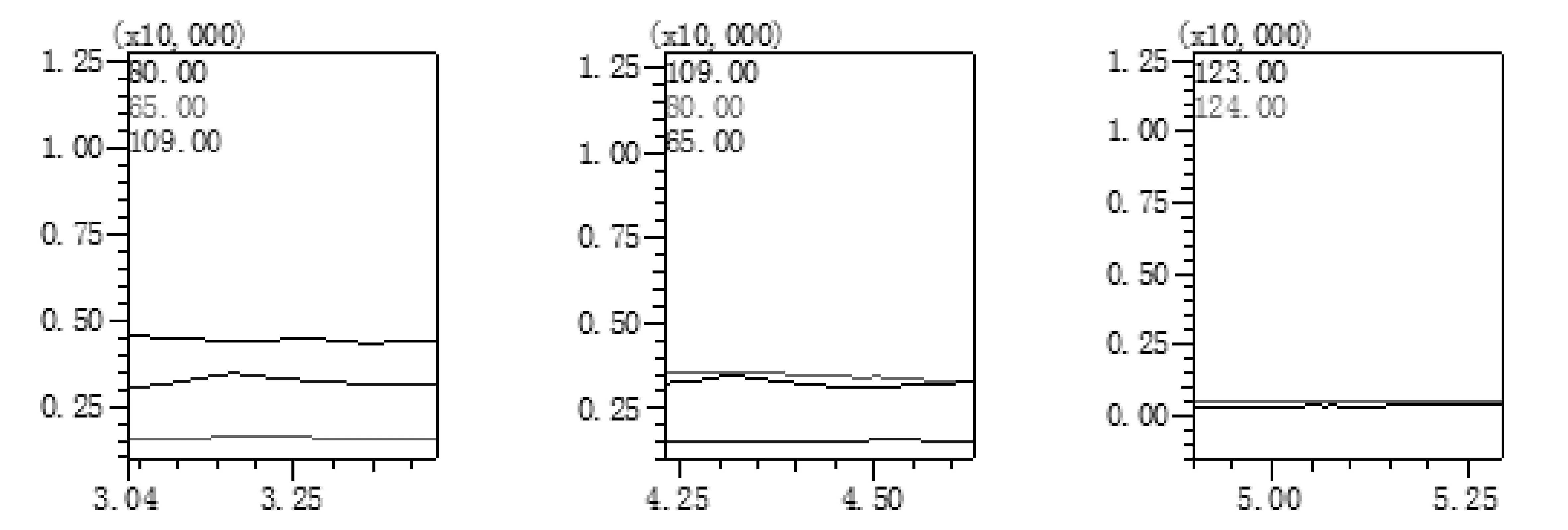
图2 样品制备液GC-MS/SIM选择离子流图
2.3 色谱条件选择
研究了起始柱温对分离效果的影响。结果表明,起始柱温对峰型、峰宽影响不大。但是,当起始柱温较低时,升温程序时间较长,目标物在色谱柱中易被分散,响应值降低;当起始柱温较高时,目标物在色谱柱中的保留时间较短,出峰时间较快,目标物间的分离度降低,且基线也不够稳定。由于甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的分子结构和理化性质均很相似,所以,色谱柱对其保留能力差异不大,保留时间相近。本方法选择起始柱温为120℃,目标物间不仅能实现有效分离,且基线平稳,检测灵敏度较高。为了尽可能提高检测分离度,在起始柱温120℃的条件下,恒温保持7min,尽量延长目标物的保留时间,使其在120℃恒温条件下能完全出峰。
分析了进样方式对检测效果的影响。为了提高检测灵敏度,采用较小的分流比进样分析,以提高目标物的响应值。研究结果表明,当采用不分流进样方式时,基线较高,且不够稳定,峰形不理想,峰宽较宽,同时有拖尾现象出现,分离度降低。当采用分流比2∶1进样分析时,上述现象均能得到很好的改善,且能满足同时检测甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的要求,因此采用分流比2∶1进样分析。图3为混合标准溶液的GC-MS/SIM总离子流图,该结果表明本方法所选择的色谱条件能够满足同时检测甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的要求。
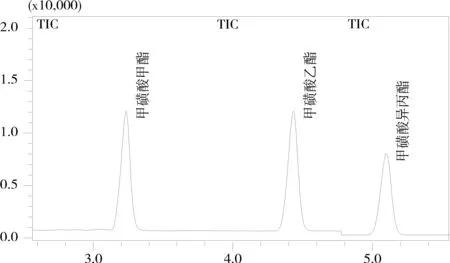
图3 混合标准溶液GC-MS/SIM总离子流图
2.4 方法的线性关系和检出限
以甲醇溶剂作为标准溶液的稀释液,精确配制目标物质量浓度均为0.01~0.40mg/L范围内的系列混合标准溶液,在上述优化的分析检测条件下,按浓度由低到高依次进行检测。以定量离子色谱峰的峰面积(Y)为纵坐标,对应的质量浓度(X,mg/L)为横坐标作图,建立标准曲线,并进行线性回归计算,得到线性方程和相关系数(R2);以定量离子色谱峰的3倍信噪比确定仪器检出限, 10倍信噪比确定仪器定量限。结果如表1所示,目标物在0.01~0.40mg/L范围内,线性关系均良好,线性相关系数均大于0.999,检出限、定量限均分别为0.003mg/L、0.01mg/L。上述结果表明,本方法适用于甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的定量分析。
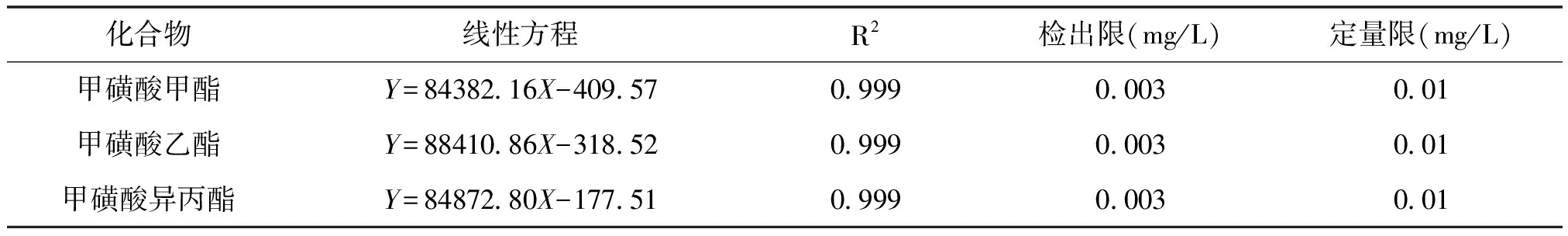
表1 方法的线性方程、相关系数(R2)、检出限和定量限
2.5 加标回收率和精密度
平行制备6份样品制备液,连续进样分析,结果如图2所示,表明,该药物中未能检测出甲磺酸酯类物质。精确配制甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯质量浓度分别均为0.16、0.20、0.24mg/L的混合标准溶液,按1.3制得样品加标制备液,进行加标回收率和精密度实验,每个加标浓度平行测定6次。结果如表2所示,目标物3个加标浓度的平均回收率为88.43%~104.15%,相对标准偏差为0.97%~1.82%。上述结果表明,本方法能满足该药物中甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的检测要求。
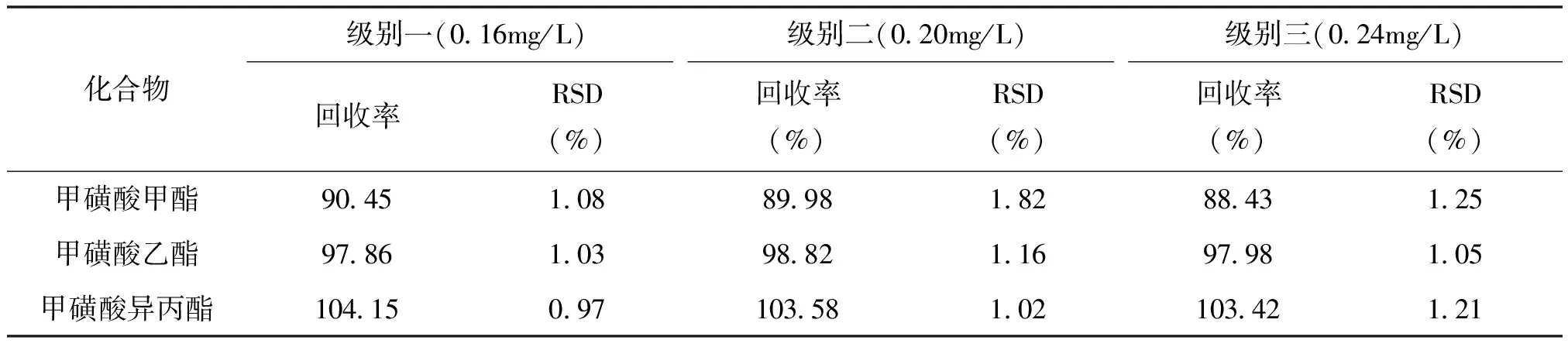
表2 方法的回收率和精密度(n=6)
3 结论
本方法建立了气相色谱-质谱-选择离子监测模式(GC-MS/SIM)检测盐酸鲁拉西酮片中甲磺酸酯类物质的分析方法。方法准确、可靠、灵敏度高,可同时测定该药物中甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的残留量,为药物中磺酸酯类物质残留的检测分析提供了有效的指导和借鉴。
[1] Glowienke S, Frieauff W, Allmendinger T, et al. Structure-activity considerations and in vitro approaches to assess the genotoxicity of 19 methane-, benzene- and toluenesulfonic acid esters[J]. Mutation Research, 2005, 581: 23-34.
[2] Dobo K L, Greene N, Cyr M O, et al. The application of structure-based assessment to support safety and chemistry diligence to manage genotoxic impurities in active pharmaceutical ingredients during drug development[J]. Regulatory Toxicology and Pharmacology, 2006, 44: 282-293.
[3]Guo T, Shi Y , Zheng L, et al. Rapid and simultaneuous determination of sulfonate ester genotoxic impuriries in drug substance by liquid chromatography coupled to tandem mass spectrometry: comparision of different ionization modes[J]. Journal of Chromatography A, 2014, 1355: 73-79.
[4]Donovan P, Smith G. Mutagenicity of N-ethyl-N-nitrosourea, N-methyl-N-nitrosourea, methyl methanesulfonate and ehyl methanesulfonate in the developing syrian hamster feuts[J]. Mutation Research,2010,699:55-57.
[5]Kaiser G S, Germann S M, Westergaard T, et al. Phenylbutyrate inhibits homologous recombination induced by camptothecin and methyl methanesulfonate[J]. Mutation Research, 2011, 713: 64-75.
[6] Walker V E, Casciano D A, Tweats D J. The viracept-ems case: impact and outlook[J]. Toxicology Letters, 2009, 190: 333-339.
[7] Ho T D, Yehl P M, Chetwyn N P, et al. Determination of trace level genotoxic impurities in small molecule drug substances using conventional headspace gas chromatography with contemporary ionic liquid diluents and electron capture detection[J]. Journal of Chromatography A, 2014, 1361: 217-228.
[8] Li W. Trace analysis of residual methyl methanesulfonate, ethyl methanesulfonate and isopropyl methanesulfonate in pharmaceuticals by capillary gas chromatography with flame ionization detection[J]. Journal of Chromatography A, 2004, 1046: 297-301.
[9] Colon I, Richoll S M. Determination of methyl and ethyl esters of methanesulfonic benzensulfonic and p-toluenesulfonic acids in active pharmaceutical ingredients by solid-phase microextraction(SPME) coupled to GC/SIM-MS[J].Journal of Pharmaceutical and Biomedical Analysis, 2005, 39: 477-485.
[10] Ramakrishna K, Raman N V V S S, Narayana Rao K M V, et al. Development and validation of GC-MS method for the determination of methyl methanesulfonate and ethyl methanesulfonate in imatinib mesylate[J]. Journal of Pharmaceutical and Biomedical Analysis, 2008, 46: 780-783.
[11] Elder D P, Teasdale A, Lipczynski A M. Control and analysis of alkyl esters of alkyl and aryl sulfonic acids in novel active pharmaceutical ingredients(APIs)[J]. Journal of Pharmaceutical and Biomedical Anslysis, 2008, 46: 1-8.
[12] Montesano M A, Whitehead Jr R D, Jayatilaka N K , et al. Measurement of ethyl methanesulfonate in human plasma and breast milk samples using high-performance liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry [J]. Journal of Pharmaceutical and Biomedical Analysis, 2010, 52: 260-264.
[13] Kakadiya P R, Reddy B P, Singh V, et al. Low level determinations of methyl mehanesulfonate and ethyl methanesulfonate impurities in lopinavir and ritonavir active pharmaceutical ingredients by LC/MS/MS using electrospray ionization[J]. Journal of Pharmaceutical and Biomedical Analysis, 2011, 55: 379-384.
