PAPASH 综合征一例研究
2018-03-15毛子慧张江安于建斌马骞曹瑞祥赵蕾李昕陈莹莹程灵云
毛子慧 张江安 于建斌 马骞 曹瑞祥 赵蕾 李昕 陈莹莹 程灵云
450052郑州大学第一附属医院皮肤科(毛子慧、张江安、于建斌、曹瑞祥、赵蕾、李昕、陈莹莹、程灵云),遗传与产前诊断中心(马骞)
PAPASH综合征(pyoderma gangrenosum,acne,pyogenic arthritis,and suppurative hidradenitis syndrome)是一种与PSTPIP1基因突变有关的自身炎症性疾病,表现为化脓性关节炎、坏疽性脓皮病、痤疮、化脓性汗腺炎[1],非常罕见。河南省医学情报研究所科技查新报告示国内发表文献中未检出中国人患化脓性关节炎、坏疽性脓皮病、痤疮、化脓性汗腺炎(PAPASH)综合征临床病例的文献报告。现将我们所见PAPASH综合征1例报道如下。
病历资料
患者男,22岁,因面颈部、背部、腋窝、腹股沟暗红色丘疹、结节、脓疱、囊肿6年,双下肢多发紫红斑块伴溃疡1年,于2016年10月31日就诊于郑州大学第一附属医院皮肤科。患者6年前无明显诱因背部出现多数炎性丘疹、脓疱,后扩展至颜面部,形成大小不等的暗红色结节,当地医院考虑痤疮未予治疗。1年前双下肢无明显诱因出现散在暗紫红色斑块,破溃融合成较大溃疡,有脓性分泌物,疼痛明显。至当地医院就诊,考虑为蜂窝织炎,给予抗生素静脉滴注及外用药物(具体用药不详),效果欠佳。为求进一步诊治来我院,门诊以“PAPASH综合征?”收入院。发病以来,精神尚好,饮食、睡眠正常,二便正常。2002年5月9日患者因发热1年余,左膝关节肿痛半年曾就诊于郑州大学第一附属医院儿科,住院检查示类风湿因子阴性,左膝关节腔积液涂片:中性粒细胞、少量淋巴及单核细胞,未见异常细胞,出院诊断为“左膝关节化脓性关节炎”。患者否认家族中类似疾病史,否认外伤史。
体检:一般情况欠佳,行走困难。体温37.0℃,脉搏76次,呼吸 19次,血压142/92 mmHg(1 mmHg=0.133 kPa)。颈部、腹股沟少数肿大淋巴结,无黏连,伴轻微触痛。其余各系统检查未见异常。皮肤科检查:面颈部、背部可见炎性丘疹、脓疱、脓肿及囊肿,部分破溃,可见凹陷性瘢痕及少量增生性瘢痕,部分有色素沉着,颈部及背部多发(图1A,1B)。双下肢多发紫红色斑块,中央坏死形成溃疡,基底凹凸不平,境界清楚,边缘皮肤呈紫红色肿胀,溃疡中心可见筛状瘢痕,部分可见黑褐色痂皮及坏死组织,挤压可见脓性分泌物(图2A)。腋窝、腹股沟可见少量硬性暗红色结节,部分破溃、溢脓(图3A,3B)。双下肢其余部位及腹股沟处可见溃疡愈合后遗留的棕褐色色素沉着。双足趾甲板可见横形分界线,将甲分为两部分,近心端为正常甲,远心端甲板变黄,表面粗糙,凹凸不平,失去光泽。
实验室及皮损组织病理检查
实验室检查:白细胞5.2×109/L,红细胞3.44×1012/L,血红蛋白121.0 g/L,血小板317× 109/L,C反应蛋白8.96mg/L(参考值<5 mg/L),性激素PROG 0.18 ng/ml(0.2~1.4 ng/ml)。尿常规、粪常规、血生化、红细胞沉降率、凝血指标均正常。抗核抗体、抗ENA谱、抗dsDNA抗体、类风湿因子、抗环瓜氨酸肽抗体、抗角蛋白抗体、抗核周因子抗体均阴性,HLA⁃B27阴性。下肢溃疡分泌物细菌培养:肺炎克雷伯菌,真菌培养阴性,抗酸染色阴性。骶髂关节、双膝关节MRI平扫:双髋关节、膝关节少量积液,双侧腹股沟区多发小淋巴结影,双膝关节内侧半月板后角变性。心电图、胸部CT及腹部彩超均未见异常。
下肢溃疡边缘皮损组织病理:表皮破溃,细胞间水肿,可见中性粒细胞进入。真皮浅中层明显水肿,局灶红细胞外溢,中性粒细胞、淋巴细胞、组织细胞弥漫性浸润,偶见嗜酸性粒细胞及小血管扩张,未见血管炎改变(图4~7)。
基因检测
应用PCR⁃Sanger测序的方法,检测了患者PSTPIP1基因(NM⁃003978.4)外显子及内含子区域的序列变异情况,检测结果:外显子区未见突变,内含子区发现c.36+68 G>A、c.137+47 G>C、c.562+114 C>G het(图8),均为杂合突变。在100例正常人该基因的测序中有45例出现了相同的突变位点。数据库分析显示均为多态性位点。
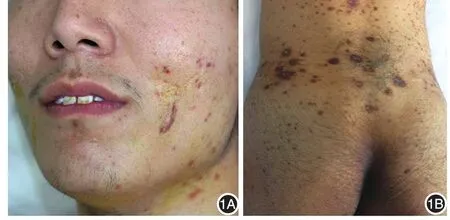
图1 患者颜面(1A)、腰背部(1B)炎性丘疹、脓疱、脓肿及囊肿,部分破溃
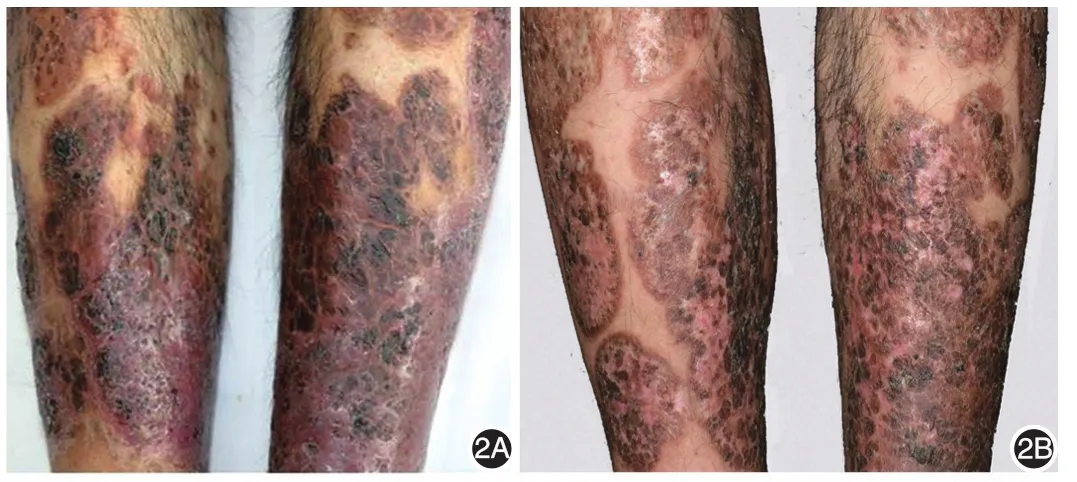
图2 治疗前后皮损表现 2A:双下肢紫红色斑块,中央坏死形成溃疡结痂,境界清楚,溃疡中心可见筛状瘢痕、黑褐色痂皮;2B:治疗10个月,双下肢皮损明显好转

图3 腋窝(3A)及腹股沟(3B)化脓性汗腺炎损害
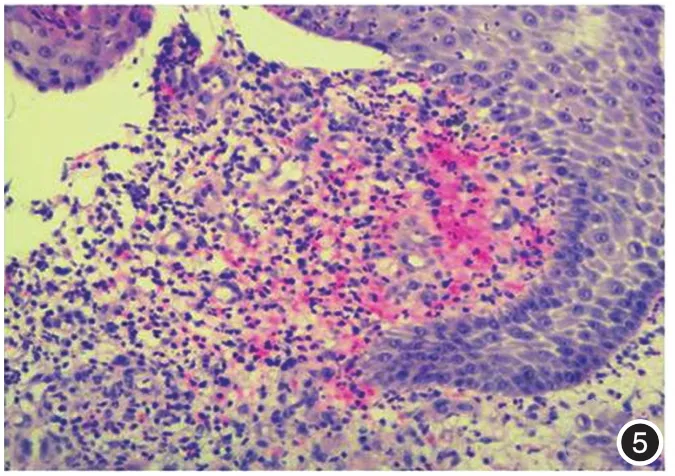
图5 真皮浅层中性粒细胞、淋巴细胞及组织细胞弥漫浸润,可见红细胞外溢(HE×200)

图6 表皮细胞间及真皮水肿,中性粒细胞进入表皮。真皮中性粒细胞浸润明显(HE×400)

图7 真皮小血管扩张,中性粒细胞、淋巴细胞及组织细胞弥漫性浸润。可见少数嗜酸性粒细胞(HE×200)

图8 患者基因检测结果 PSTPIP1基因内含子存在杂合突变。箭头所示为突变位点
由于PAPASH综合征病例过少,虽然本例患者PSTPIP1基因未发现明确或疑似致病突变,但结合本例患者的临床特征、实验室检查、组织病理结果,最终诊断:PAPASH综合征。
治疗经过
2016年11月入院后给予甲泼尼龙30 mg、头孢米诺2 g每天2次静脉滴注;异维A酸软胶囊20 mg每天1次、沙利度胺50 mg每晚1次口服,生理氯化钠溶液冷湿敷患处,早晚各1次,每次30分钟,溃疡处红光治疗每天1次,每次20分钟,同时予护胃、补钾、补钙等辅助治疗。患者经上述治疗13 d,双下肢溃疡明显好转,甲泼尼龙减量至25 mg,余用药方法同前,复查血尿常规、血生化未见异常后出院。出院后继续予泼尼松30 mg早8点口服,异维A酸软胶囊20 mg每日1次口服,沙利度胺50 mg每晚1次口服,夫西地酸、姜黄消痤擦剂外用,兼以补钙、补钾、护胃等辅助治疗。2017年8月复诊,痤疮、化脓性汗腺炎皮损已完全消退,双下肢坏疽性脓皮病溃疡性皮损基本愈合,部分遗留萎缩性瘢痕及褐色薄痂,部分恢复、接近正常皮肤(图2B)。治疗调整为泼尼松10 mg早8时口服,沙利度胺50 mg每晚1次口服,巩固治疗。现仍在随访中。
讨 论
PAPASH综合征由Marzano等[1]于2013年首次报道,1例16岁女性患者,曾患有非轴向型关节炎,关节腔积液示中性粒细胞炎症,化脓性汗腺炎、轻度痤疮2年,坏疽性脓皮病6个月,口服甲泼尼龙50 mg/d和局部外用他克莫司软膏(0.1%,每日2次)后,坏疽性脓皮病缓解,基因检测结果PSTPIP1基因的外显子10和11的c.831G→T核苷酸取代,导致p.E277D错义突变。PSTPIP1基因突变,导致其编码的脯氨酸/丝氨酸/苏氨酸磷酸酶相互作用蛋白(CD2结合蛋白1)与pyrin的亲和力增加,从而使caspase⁃1活性的上调和IL⁃1的活化,产生中性粒细胞介导的反应[2]。
2014年,Garzorz等[3]报道1例39岁女性患者,坏疽性脓皮病3年,既往有银屑病病史、严重的化脓性汗腺炎及面部轻度寻常痤疮,应用环孢素治疗有效,但减量过程中出现化脓性关节炎恶化及背部和手关节的疼痛,类风湿因子阴性,骨扫描显示血清阴性关节炎,在PSTPIP1基因的遗传分析中发现了新的突变(具体突变位点文中未指出),因此在对比了PAPA和PASH综合征后,作者用PAPASH综合征来定义这一疾病,并提出这一疾病的关键分子特征是Th⁃17/TNF⁃α轴的中性粒细胞活化。Ursani等[4]于2016年报道了1例44岁男性患者,既往病史包括痤疮、化脓性汗腺炎、慢性腹泻、复发性皮肤溃疡及双侧膝、踝、指关节的关节炎,下肢溃疡活检提示坏疽性脓皮病,结肠镜显示溃疡和瘘形成,溃疡性病变的活组织检查提示溃疡性结肠炎,根据典型的临床表现及既往病史,诊断为PAPASH综合征,该病例未行基因检测。
由此可见,PAPASH综合征是一种罕见的类似于PAPA和PASH综合征的自身炎症性疾病,国外仅报道3例,国内未见报道。据国外报道可知,目前该综合征并无明确定义,其诊断主要依靠典型的临床表现及既往病史,其真正病因未知。结合相似疾病PAPA综合征的研究,部分病例PSTPIP基因检测发现了新的突变位点,从而推测该疾病可能与PSTPIP1基因的突变有关。在应用PAPASH综合征命名这一疾病时,PSTPIP1基因突变并非必要条件,更多是依据临床表现归类于与PAPA综合征为同一疾病谱的自身炎症性疾病中[5],正如该疾病谱中的PASH、PASS综合征(坏疽性脓皮病、囊肿型痤疮、化脓性汗腺炎和轴向型脊柱关节炎)都是在PAPA综合征的基础上定义的。PAPA综合征由Lindor等[6]于1997年首次报道,表现为化脓性关节炎,坏疽性脓皮病和痤疮,是由染色体15q上的PSTPIP1/CD2BP1基因突变引起的常染色体显性遗传性自身炎性疾病,其分子特征为IL⁃1β的过量产生。PASH综合征由Markus等于2012年首次报道[7],表现为坏疽性脓皮病,痤疮和化脓性汗腺炎,不存在关节累及,未发现PSTPIP1基因本身的突变,而是与PSTPIP1启动子GTCC区域中的微卫星重复序列的数目增加有关。PAPA、PASH、PAPASH综合征的鉴别诊断[8]主要集中在特征性临床表现和基因突变方面,由于三者共属于自身炎症性疾病,故其病理生理及治疗方案大致相同。
本例患者依据长期痤疮、化脓性汗腺炎病史,典型坏疽性脓皮病及既往化脓性关节炎病史,PAPASH综合征诊断明确。患者的PSTPIP1基因测序结果示外显子区未发现突变位点,但内含子发生了复合杂合突变,初步分析突变位点属多态性位点,故该突变代表的意义及致病性尚待进一步明确。γ分泌酶基因的功能性缺失突变在PASH综合征研究中已有报道[9],国外文献仅对PAPASH综合征PSTPIP1基因进行检测,并未检测γ分泌酶基因及其亚基,有必要进一步明确。虽然PSTPIP1基因突变可以在多种坏疽性脓皮病相关的综合征中检出,但部分病例也未发现突变,且突变位点也不尽相同[10]。本例患者治疗主要针对坏疽性脓皮病和痤疮,给予糖皮质激素、抗生素及沙利度胺治疗后,双下肢溃疡逐渐愈合。由于本例患者痤疮表现更为严重,化脓性汗腺炎相对较轻,所以在治疗上更倾向于囊肿型痤疮的治疗方案,选异维A酸而未选用阿维A。随访中糖皮质激素逐步减量,皮损逐渐好转愈合,显示上述针对性治疗有效。
[1]Marzano AV,Trevisan V,Gattorno M,et al.Pyogenic arthritis,pyoderma gangrenosum,acne,and hidradenitissuppurativa(PAPASH):a new autoinflammatory syndrome associated with a novel mutation of the PSTPIP1 gene[J].JAMA Dermatol,2013,149(6):762⁃764.doi:10.1001/jamadermatol.2013.2907.
[2]Smith EJ,Allantaz F,Bennett L,et al.Clinical,molecular,and genetic characteristics of PAPA syndrome:a review[J].Curr Genomics, 2010,11 (7):519 ⁃527. doi: 10.2174/138920210793175921.
[3]Garzorz N,Papanagiotou V,Atenhan A,et al.Pyoderma gangrenosum, acne, psoriasis, arthritis and suppurative hidradenitis(PAPASH)⁃syndrome:a new entity within the spectrum of autoinflammatory syndromes?[J].J Eur Acad Dermatol Venereol,2016,30(1):141 ⁃143.doi:10.1111/jdv.12631.
[4]Ursani MA,Appleyard J,Whiteru O.Pyogenic arthritis,pyo⁃derma gangrenosum,acne,suppurative hidradenitis(PA⁃PASH)syndrome:an atypical presentation of a rare syndrome[J].Am J Case Rep,2016,17:587⁃591.
[5]Gönül M,Cevirgen CB,Keseroglu HO,et al.New described dermatological disorders[J].Biomed Res Int,2014,2014:616973.doi:10.1155/2014/616973.
[6]Lindor NM,Arsenault TM,Solomon H,et al.A new autosomal dominantdisorderofpyogenic sterile arthritis,pyoderma gangrenosum,and acne:PAPA syndrome[J].Mayo Clin Proc,1997,72(7):611⁃615.doi:10.1016/S0025⁃6196(11)63565⁃9.
[7]Braun⁃Falco M,Kovnerystyy O,Lohse P,et al.Pyoderma gangrenosum,acne,and suppurative hidradenitis(PASH):a new autoinflammatory syndrome distinct from PAPA syndrome[J].J Am Acad Dermatol,2012,66(3):409 ⁃415.doi:10.1016/j.jaad.2010.12.025.
[8]Cugno M,Borghi A,Marzano AV.PAPA,PASH and PAPASH syndromes:pathophysiology,presentation and treatment[J].Am J Clin Dermatol,2017,18(4):555⁃562.doi:10.1007/s40257⁃017⁃0265⁃1.
[9]Calderón⁃Castrat X,Bancalari⁃Díaz D,Román⁃Curto C,et al.PSTPIP1 gene mutation in a pyoderma gangrenosum,acne and suppurative hidradenitis(PASH)syndrome[J].Br J Dermatol,2016,175(1):194⁃198.doi:10.1111/bjd.14383.
[10]Gasparic J,Theut RP,Jemec GB.Recognizing syndromic hidradenitis suppurativa:a review of the literature[J].J Eur Acad Dermatol Venereol,2017.doi:10.1111/jdv.14464.
