基于关键功能模块挖掘的蛋白质功能预测
2018-03-10赵碧海李学勇田清龙杨品红
赵碧海 李学勇 胡 赛 张 帆 田清龙 杨品红 刘 臻
由于蛋白质在不同生物过程中扮演重要角色,注释功能未知的蛋白质是后基因时代的重要任务之一.生物实验确定蛋白质功能存在耗时多和费用高的问题[1].因此,基于计算的功能预测成为非常重要的替代方法.然而,这种方法需要准确而可靠的自动功能预测器.现有的基于计算的功能预测方法都是建立在数据库中已经注释的蛋白质的功能之上.虽然相互作用数据、序列数据和蛋白质结构数据等都已用于蛋白质功能预测算法,但是设计一种有效的方法充分利用各种不同的生物信息依然是一个巨大的挑战,源于这些生物数据的异构性、复杂性和多样性.根据整合这些不同数据源的方式不同,这些基于计算的预测方法可以分为四类:基于多特征向量方法、基于多分类器方法、基于核的方法和基于网络的方法.网络是一种很好的描述蛋白质之间关系的途径,而且大量基于网络的方法为我们提供了有效的工具从网络中挖掘信息,这也有助于我们理解细胞生命活性物的复杂机制.
大部分基于网络的蛋白质功能预测方法都是从蛋白质相互作用(Protein-protein interaction,PPI)网络提取信息.这些方法都建立在一个发现的基础上:大约70%∼80%的蛋白质与它们在PPI网络的相互作用伙伴至少共享一项功能[2].一些方法通过PPI网络中的直接或间接邻居节点预测未注释的蛋白质功能.上述的这些方法独立地为每一个蛋白质预测功能.还有一些方法将PPI网络中的蛋白质分成多个功能模块,并为相同的模块注释相同的功能[3].这类方法聚类形成模块或复合物的方式存在差异.由于相互作用数据中存在假阳性和假阴性,一些研究者结合相互作用网络和异构生物数据,提高功能预测的准确率,例如基因表达数据[4]、同源数据[5]、蛋白质复合物数据[6]、结构域数据[7]等.
另一种流行的基于网络并利用生物信息资源的方法是基于GO term的功能相似性建立功能关联网络.蛋白质功能描述为结构化的标准词汇,并存储在基因本体数据库.GO term之间的父亲–孩子关系可以表达为有向无环图.考虑到两个相似的功能共同注释一个共同的蛋白质以及两个相互作用的蛋白质倾向于共享同一功能,一些研究者结合PPI网络和功能相似性,从而提高功能预测的准确率.由于PPI网络的不完整性,其他的异构数据也被整合进来.Peng等[8]结合PPI网络和Domain信息,利用蛋白质的功能相似性,提出名为DCS的蛋白质功能预测方法.进一步,加入蛋白质复合物信息,提出了改进的DSCP方法.大部分整合异构数据的方法基本采取如下思路:1)生成各种功能相关网络(每一个数据源对应一个或多个网络);2)这些单独的网络通过加权汇总的方式形成一个复合网络.这些方法的区别在于单个网络形成复合网络时,不同方法权值比例和优化方式存在差异.
综上所述,整合多元生物数据能够有效弥补相互作用网络不完整性和噪声的问题,提高基于网络的蛋白质功能预测方法的准确率.但是,引入其他生物信息后,使得蛋白质之间的联系更加复杂,更加多元化.现有的方法基本都采取合并多种类型的相互作用的处理方式,这虽然能够一定程度增加正确匹配的功能数量,但也会同时引入更多的噪声功能,最终使得整体预测性能提升不大.上述提及的某些方法先构建多种功能关联网络,然后再采取加权汇总的方式将多个单独网络构成一个复合网络.不同网络在加权汇总时的比重各不相同,而每个网络的比重参数成为影响功能预测方法的重要因素.参数的设置一般会根据经验值设置.即便是通过优化的方式获取,也存在不同数据集有不同设置的问题.从这些问题出发,本文在原有研究基础之上,结合PPI网络、蛋白质复合物数据和蛋白质结构域数据建立多关系网络.考虑到蛋白质功能与模块之间的紧密联系,提出一种基于多关系网络中关键功能模块挖掘的蛋白质功能预测方法(Prediction of functions based on essential functional modules mining from a multi-relational network,PEFM).蛋白质的功能不是由单个蛋白质独立完成,而是与其他蛋白质相互作用共同执行机体功能,蛋白质功能与功能模块之间存在紧密联系.关键功能模块是指相互间紧密联系的蛋白质组成的功能模块或复合物.移除关键功能模块会使得生物体丧失许多重要分子功能.因此,通过挖掘关键功能模块有助于提高蛋白质功能预测算法的准确率.PEFM方法依次遍历多关系网络分解得到的每一个简单网络,挖掘高内聚、低耦合的稠密子图形成不同网络层次的关键功能模块集合.模块中节点的全部功能用于注释测试蛋白质.多个数据集的实验结果验证了PEFM算法的有效性.
1 PEFM算法
细胞功能不是由单个蛋白质完成,而是通过多个紧密联系的蛋白质构成模块,共同执行.蛋白质功能与模块之间存在紧密联系,模块划分为蛋白质功能预测提供了途径.本文通过聚类,形成高内聚、低耦合的功能模块,进而实现蛋白质功能预测.
1.1 多关系网络构建
受实验条件限制,高通量方法获得的蛋白质相互作用数据具有不完整性,限制了蛋白质功能预测算法的性能.结合多元的生物信息和蛋白质相互作用网络,降低相互作用数据的实验错误带来的负面影响,是当今基于相互作用网络的功能预测算法的发展趋势.多元异构数据包括基于时间序列的基因表达信息、蛋白质结构域信息、复合物信息、亚细胞定位信息等.在原有研究基础之上,本文结合蛋白质相互作用网络的拓扑特性、蛋白质结构域信息和蛋白质复合物信息构建适合功能预测的多关系网络[7].相比之前构建的研究基础,本文在建立多关系网络时,增加了蛋白质复合物数据.由于实验方法获得的蛋白质相互作用数据和结构域数据存在假阴性,存在某些蛋白质执行共同功能,却没有在前期构建的网络中体现的情况.通过融入复合物数据,能够为更多的蛋白质预测功能.
蛋白质结构域是分子的一个特别区域,具有独立的功能.有的蛋白质仅仅包含一个结构域,有的蛋白质可能包含多个不同类型的结构域.一个结构域也可能出现在多个不同的蛋白质当中.蛋白质的新功能常常利用结构域重组完成.蛋白质执行生物功能离不开结构域,由此可见,蛋白质功能与结构域之间存在紧密的联系.学者们开始尝试利用结构域信息,提高功能预测算法的准确率.
本文首先针对蛋白质结构域信息与蛋白质功能之间的关联开展统计分析.本次实验选定的蛋白质相互作用网络包含5093个蛋白质,其中具有功能注释的蛋白质数量是2894,至少包含1个结构域的蛋白质数量是3056,既有功能注释又有包含结构域的蛋白质数量为1887个,如图1所示.从图1不难看出,具有结构域的蛋白质中,61.75%的蛋白质至少具有1项功能;2894个被注释的蛋白质中,有65.2%的蛋白质包含结构域.
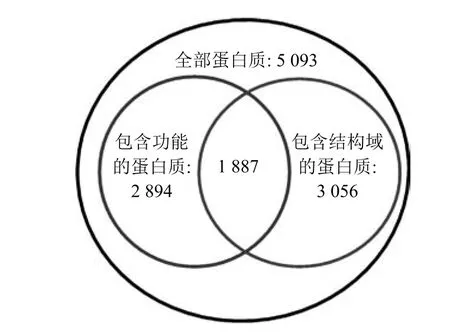
图1 结构域与蛋白质功能关系综合统计Fig.1 Statistics of relationship between domains and protein functions
进一步地,本文统计分析2894个功能已知的蛋白质之间共享功能和共享结构域的情况,其中42%的蛋白质与其他蛋白质共享功能的同时还共享相同的结构域.表1详细列出了蛋白质功能数量分布与共享结构域之间的关系.
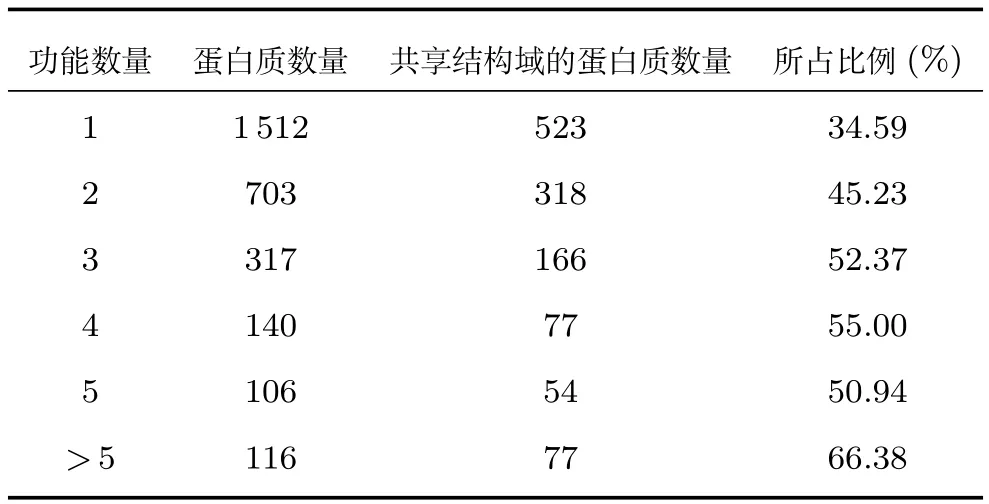
表1 蛋白质功能数量统计Table 1 Quantity statistics of protein functions
从表1不难看出,1512个蛋白质仅有1项功能,其中34.59%的蛋白质与其他蛋白质共享功能的同时还共享结构域.而当功能数量增多时,共享结构域的蛋白质比例明显增高.由此可见,蛋白质间共享结构域的特性有助于提升蛋白质功能预测性能,尤其适用于功能数量较多的蛋白质.
本文构建的多关系网络中,蛋白质之间相互作用的第一种类型为共享结构域.为提高预测的性能,我们依据上述的统计分析结论对该种类型的相互作用加权.统计表明,两个蛋白质包含相同结构域的比例越高,它们之间存在联系的可能性越大.本文提出的PEFM算法中,若两个蛋白质包含共同类型的结构域,则它们之间存在相互作用.相互作用的权值通过共同结构域的数量所占比重刻画,加权计算方式如下:
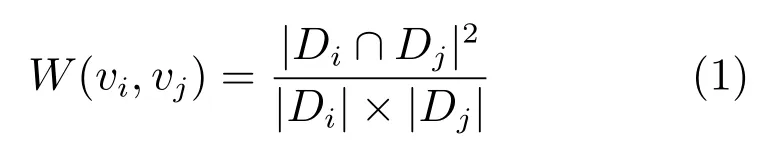
其中,W(vi,vj)表示蛋白质vi和vj共享结构域的可能性.Di和Dj分别表示蛋白质vi和vj的不同类型结构域构成的集合,Di∩Dj是两个蛋白质相同结构域类型构成的集合.若Di或Dj为空集,则权值简单地设置为0.
蛋白质复合物由多个紧密联系的蛋白质组成,并共同执行某些生物功能.很多蛋白质只有聚合成复合物,并与其他蛋白质相互作用才能体现出某种功能,由此可见,蛋白质复合物与功能之间存在紧密联系.图2显示了蛋白质功能数量与共享蛋白质复合物之间的关系.
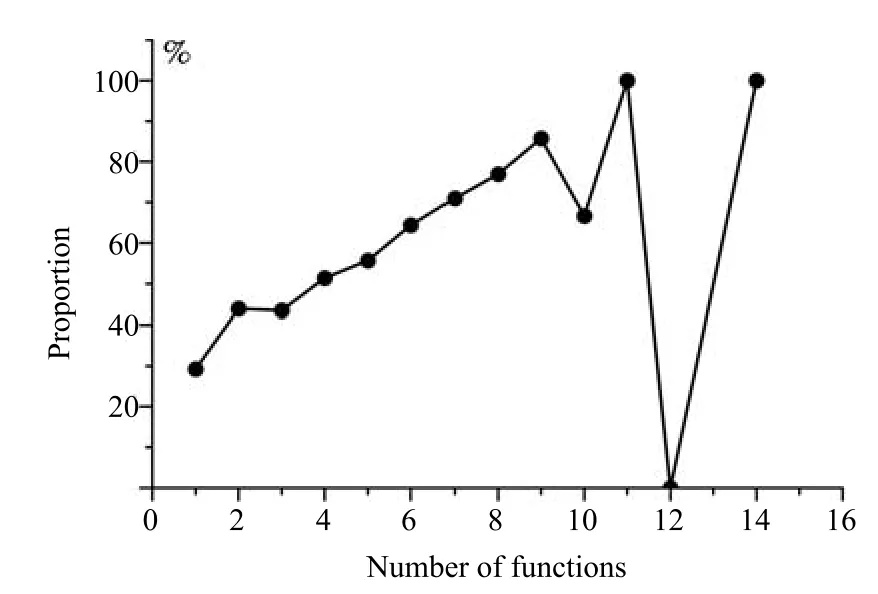
图2 蛋白质功能与共享复合物统计分析Fig.2 Statistics of relationship between proteincomplexes and functions
从图2可以看出,对于仅包含1项功能的蛋白质,30%左右的蛋白质与其他有功能注释的蛋白质包含在相同的复合物中.随着功能数量的增多,这个比例明显增高.包含12项功能的蛋白质仅1个,此时比例为0,可以认为是偶然事件.两个蛋白质共享相同的复合物是本文构建的多关系网络中的第二种类型.这种类型的相互作用加权类似于第一种类型.
对于网络中的两个蛋白质vi和vj,Ci和Cj分别表示包含vi和vj的复合物组成的集合,共享复合物的相互作用加权计算方式如下所示:
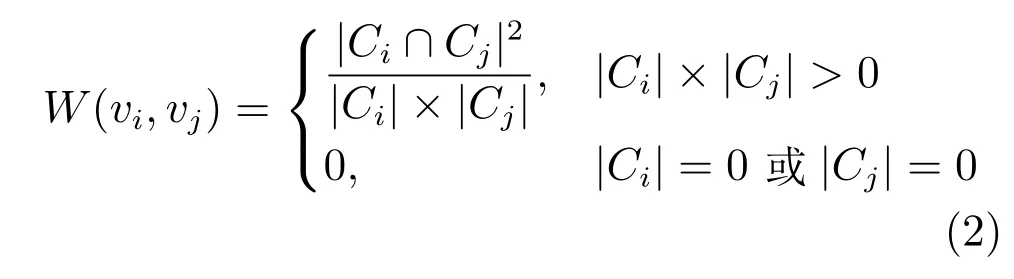
其中,Ci∩Cj表示同时包含vi和vj的复合物形成的集合.若vi或vj没有出现在任何复合物中,则W(vi,vj)=0.
多关系网络中的最后一种类型来源于相互作用网络拓扑特性的分析.众所周知,蛋白质相互作用网络具有小世界特性和稀疏性,且存在假阳性.如果两个蛋白质都同时与第三个蛋白质发生相互作用,则这两个蛋白质间相互作用假阳性的可能性比较小,共同参与模块执行相同功能的可能性比较大.因此,一对蛋白质之间相互作用的概率可以通过他们共有的邻居节点数量确定.本文采用ECC计算蛋白质之间连接的权值.计算公式如下:

其中,Ni和Nj分别表示vi和vj的邻居集合.图3是本文结合PPI网络拓扑特性、蛋白质结构域信息和复合物信息构建的多关系网络的可视化展示.
图3中,第一层表示蛋白质间因为隶属同一复合物而发生相互作用,第二层表示蛋白质间因为包含共同的结构域而相互作用,第三层则是在相互作用网络的基础上,通过拓扑特征分析建立.图中虚线将各层相同的蛋白质相连,也就是说三层包含相同的蛋白质集合,不同的是蛋白质间的相互作用.
1.2 关键功能模块挖掘
细胞的功能是由多个紧密联系的蛋白质通过形成功能模块执行.Zotenko等提出关键复合物生物模块(Essential complex biological modules,ECOBIMs)[9],它是一组紧密联系且共享生物功能的蛋白质组成.Nepusz等[10]指出,子图能够表示为复合物应该满足两点:1)子图内包含许多可靠的相互作用;2)子图能够与网络的剩余部分很好地区分.受此启发,考虑到蛋白质功能与模块之间的紧密联系,本文通过从多关系网络中挖掘关键功能模块,实现蛋白质功能预测.在介绍关键功能模块挖掘算法前,先简要介绍算法所涉及的几个定义.
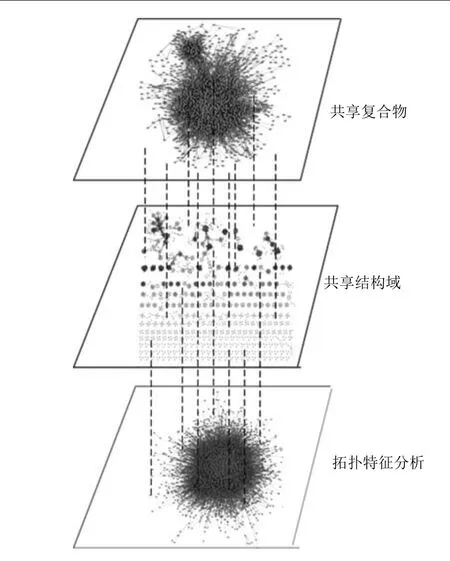
图3 多关系网络可视化显示Fig.3 Visualization of a multi-relationship network
定义1.加权度(Weighted degree,WD).给定加权网络G=(V,E,W),节点u∈V,V={v1,v2,···,vn},E={e1,e2,···,em},W={w(e1),w(e2),···,w(em)},w(ei)表示边ei的权值.WD(u,G)表示u在G内的加权度,定义如下:
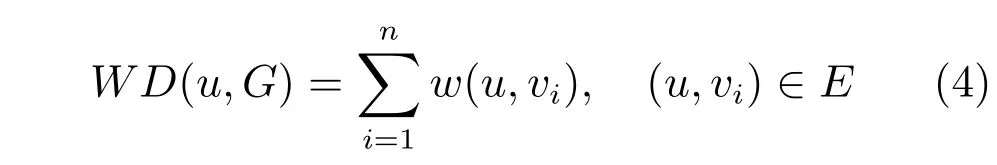
加权度描述了节点与子图之间的耦合程度.加权度越大,节点与子图内节点之间的联系越紧密.本文采用加权度描述子图与网络剩余部分的区分度.
定义2.加权稠密度(Weighted density degree,WDD).给定加权子网络G=(V,E,W),V={v1,v2,···,vn},E={e1,e2,···,em},W={w(e1),w(e2),···,w(em)},w(ei)表示边ei的权值.WDD(G)表示子网G内的加权稠密度,定义如下:
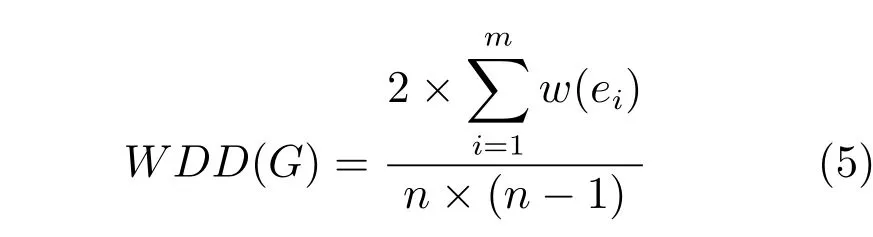
加权稠密度用以描述子图内部节点之间的连接紧密程度.本文通过加权稠密度衡量子图能否表示为高内聚的功能模块.
在PEFM方法中,若子图的加权稠密度超过给定阈值,且内部节点与子图的加权度大于节点与邻居子图的加权度,则该子图可以表示为一个高内聚、低耦合的关键功能模块.邻居子图由子图内部节点的邻居节点组成,并且这些邻居节点不出现在子图内.
关键功能模块挖掘的基本思路是:对于待注释功能的蛋白质v,PEFM算法每次遍历同种类型的相互作用,从而得到不同类型相互作用对应的关键功能模块.本文中,从v出发,通过3次遍历,最多可以得到3个关键生物模块.每次遍历时,v的邻居节点根据与v的连接紧密程度从大到小的顺序进入队列.初始的关键功能模块集合S={v},算法依次从队列中取出一个邻居节点并尝试加入集合S,若加入邻居节点后,S对应的子图加权稠密度超过设定的阈值T,则保留该节点,否则将邻居节点从S中移除,得到一个高内聚的稠密功能模块.考虑到模块中某些节点可能与外部子图存在更加紧密的联系,需要对子图S做进一步的筛选.NS是由S中所有节点的邻居节点形成的子图,若S中某一节点u在NS中加权度超过其在S中的加权度,则从S中移除u.若S的尺寸超过2个,则形成一个高内聚、低耦合的关键功能模块.我们通过一个实例描述算法在某一网络中关键功能模块的挖掘过程.如图4所示,A节点为待注释功能的测试蛋白质,加权稠密度阈值T=0.2.首先将A的邻居节点根据连接紧密程度依次放入队列Q={C,B,D,E},初始关键功能模块集合S={A}.依次从队列Q中取出节点尝试放入S中,并计算S的加权稠密度.依次将C,B,D,E放入S中后,得到的关键功能模块集合分别是{A,C},{A,C,B},{A,C,B,D}和{A,C,B,D,E},对应的加权稠密度分别是0.5,0.42,0.24,0.16.由于加入邻居节点E后,模块的加权稠密度低于设定的阈值,因此从模块中移除节点E,形成高内聚的关键功能模块集合S={A,C,B,D}.C,B和D的邻居节点形成邻居子图NS={H,F,G,K}.由于D在NS中的加权度为0.7,大于其在S中的加权度0.2,从S中移除D.最终得到关键功能模块S={A,C,B}.
以下是关键功能模块挖掘伪代码描述:


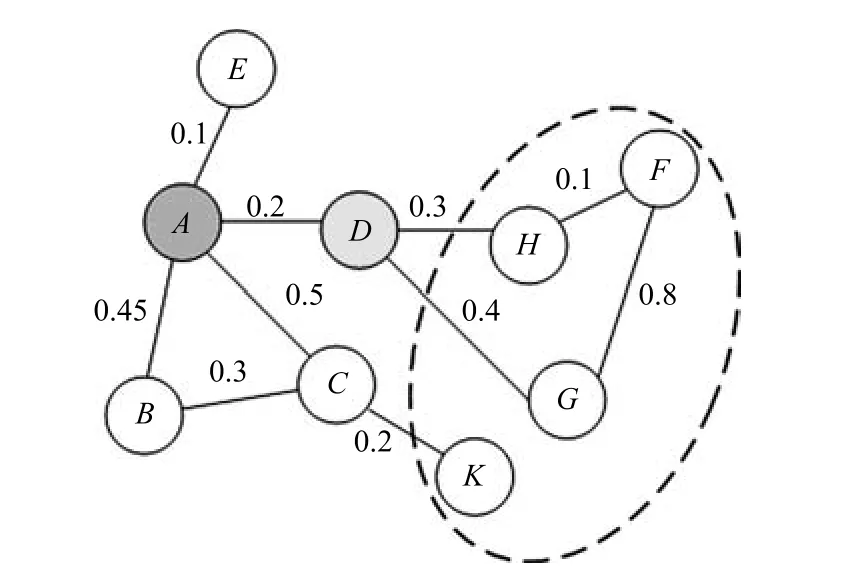
图4 关键功能模块挖掘实例Fig.4 Example of an essential functional module mining
1.3 功能预测
算法的最后一个阶段是根据挖掘的关键功能模块形成候选功能列表,并注释测试蛋白质.在上一阶段,已经产生每一种类型联系对应的关键功能模块.然而,不同类型的联系对于蛋白质功能预测的重要性各不相同.为此,我们为不同类型的联系设置不同的重要性系数.重要性系数的计算如下:

其中,P(i)表示第i种类型联系的优先级.优先级的设置源于统计分析的结果.本文分别在每种类型联系构成的简单网络上运行经典的功能预测算法–邻居计数法(Neighbour counting,NC),预测蛋白质功能,并计算每种情形下NC法的预测性能,包括敏感性、特异性和F-measure(相关定义见第2.2节),实验结果如图5所示.当NC方法运行在仅包含共享复合物类型的网络时,能获得最高的敏感性和综合性能指标F-measure.共享结构域类型的性能次之,PPI拓扑特征类型的性能最低.因此,共享复合物类型的优先级设置为1,共享结构域类型的优先级设置为2,而PPI拓扑特征类型的优先级为3.
对于功能未知的测试蛋白质u,假设挖掘的关键功能模块集合FM={fm1,fm2,fm3},fmi={pi1,pi2,···,pin}(i∈[1,3])表示第fmi个模块包含的蛋白质集合,WDD={wdd1,wdd2,wdd3}表示关键功能模块的加权稠密度,F={f1,f2,···,fm}是三个关键功能模块中所有蛋白质的全部已知功能形成的集合.对于fi中某一功能,可根据下式计算其排名得分:
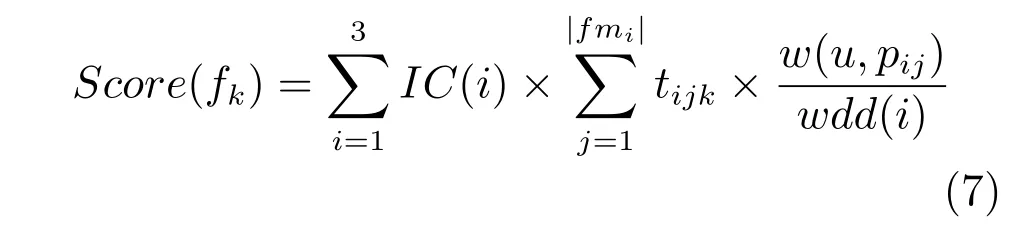
其中,w(u,pij)表示蛋白质u和pij通过第i种联系时的权值.若关键功能模块fmi内的蛋白质pij包含功能fk,则tijk=1,否则tijk=0.

图5 不同类型联系对预测的影响Fig.5 Impact of diあerent types of connection
由于预测得到的候选功能比较多,有的功能是噪声,不宜注释测试蛋白质.为此,PEFM算法将所有候选功能按照得分降序排列,然后从中选取前N项功能作为u的预测功能.N是关键功能模块中与测试蛋白质u联系最为紧密的蛋白质的功能数量.联系的紧密程度可以用相互作用的权值表示.
2 实验结果和分析
2.1 实验数据
本次实验将采用酵母蛋白质相互作用网络.因为该物种的相互作用数据和功能数据较为完整,并被用于现有的功能预测算法实验分析.我们将详细介绍和分析DIP[11]数据集的结果,也将简要分析BioGrid[12]数据集、Gavin[13]数据集和Krogan[14]数据集.DIP数据包含5093个蛋白质和24743组相互作用,Krogan数据集则包括了3672个蛋白质和14317个相互作用,Gavin数据库由1855蛋白质及7669蛋白质间相互作用组成,BioGrid包括5616个蛋白质和52833组相互作用.蛋白质功能数据为最新版本,从GO官方网站获取[15].本次实验去除了注释蛋白质数量小于10个或者大于200的功能条目,旨在提高算法的公平性.处理完毕后,注释文件包含267个不同的GO条目.下载的GO文件进行了格式转换,原始的GO文件为UniProtKB[16]格式,转换后的格式为Ensemble Genomes Protein.用于构建多关系网络的Domain数据从Pfam[17]数据库获取.Domain文件包含1107种不同类型的结构域,覆盖相互作用网络的3056个蛋白质.另一种异构数据,蛋白质复合物数据采用基准集CYC2008[18].CYC2008通过高通量的生物实验获得,由408个Benchmark复合物组成.为了检验PEFM算法的有效性和预测准确率,我们选取了FPM[7],Zhang[19],D-PIN[4],DCS[8], NC[2],PON[20]作为对比算法.本文将从多方面对比PEFM算法和竞争算法的性能.
2.2 评价指标
在测试蛋白质功能预测算法性能时,通常采用交叉验证法.蛋白质集合被划分为测试集和训练集.训练集中的蛋白质用于帮助功能预测算法实现对未知功能的蛋白质注释.测试集中蛋白质的功能被人为剥离,利用预测算法得到其预测功能.预测结束后,对比预测的功能与真实的蛋白质功能的匹配情况,从而计算功能预测算法的预测准确率.交叉验证进一步可以划分为留一法验证和留部分法验证.留一法验证是指每一轮预测时,仅保留一个功能已知的蛋白质在测试集中,剩余的蛋白质全部进入训练集.留部分法验证是指随机地选取一定比例的蛋白质放入测试集,例如10%,20%,50%.剩下的功能已知的蛋白质放入训练集.然后根据预测算法设定的功能选取策略选取一定数量的功能.算法的预测准确率由预测的功能与实际功能之间的匹配率决定.
在计算功能预测算法的预测准确率时,一般采用Speci fi city(特异性)和Sensitivity(敏感性)两种评价指标.Speci fi city主要针对预测功能集,指预测集合中被真实功能匹配的功能所占比例.Sensitivity主要针对标准集,指标准集中被预测的功能匹配的功能所占比例.Speci fi city和Sensitivity的形式化定义如下:
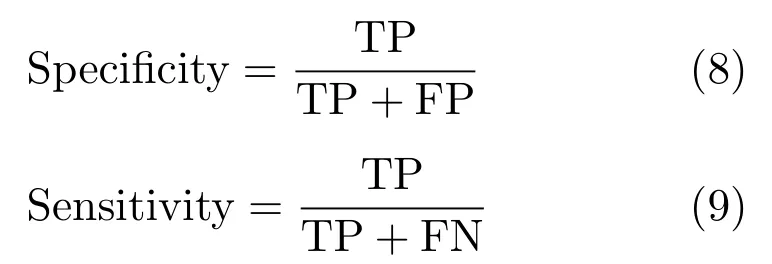
其中,TP(True positive)指预测集合中被标准集合中匹配的功能数量.FP(False positive)指预测集合中没有被任何真实功能匹配的数量.换句话说, FP等于预测的功能数量减去TP.FN(False negative)指标准集合中没有被任何预测功能匹配的真实功能数量.由于真实功能数量是固定的,在预测蛋白质功能时,提高候选功能数量,可以提高TP值,从而提高Sensitivity值.同时导致FP增长更快,导致Speci fi city明显下降.F-measure是一项综合衡量预测算法性能的指标,是Speci fi city和Sensitivity的调和平均值.
2.3 参数分析
PEFM算法中,为评估子图加权稠密度,我们引入自定义参数T.本节将分析T对算法性能的影响,并确定T的合适取值.根据定义2可知,T的取值范围在区间[0,1].图6显示了在四个数据集(DIP,Krogan,Gavin和BioGrid)上,PEFM算法的F-measure值随着T值变化的情况.
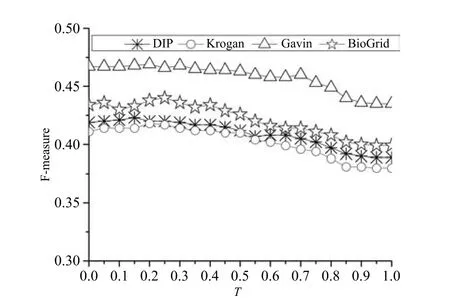
图6 参数T的影响Fig.6 The eあect of threshold T
从图6可以看出,在DIP数据集上,参数T取值0.15时,PEFM算法获得最高的F-measure值0.423.对于Krogan和Gavin数据集,T取值0.2时,综合性能指标F-measure最大,分别是0.418和0.469.对于BioGrid数据集,T=0.25时,F-measure达到最大值0.44.
2.4 留一法验证
本次实验选定的PPI网络中,共有5093个蛋白质,其中2894个蛋白质有功能注释.我们首先分析PEFM和其他六种方法对这2894个蛋白质预测功能的整体性能.图7显示了各种方法的特异性、敏感性和F-measure的平均值.2894个蛋白质中被PEFM,D-PIN,FPM,Zhang,DCS,NC和PON至少正确预测一个功能的蛋白质数量分别为1546,1506,1407,801,1118,1626和566.PEFM覆盖蛋白质数量比D-PIN,FPM,Zhang,DCS和PON分别提高2.67%,9.88%,93.01%,38.28%和173.14%.
从图7可以看出,PEFM具有最高的特异性(Speci fi city),这意味着PEFM 算法预测的功能中错误(噪声)功能所占比例最少.敏感性(Sensitivity)方面,PEFM比FPM、Zhang、DCS和PON分别提高了15.37%,95.63%,37.03%和206.7%.这说明,相比这四种功能预测算法,PEFM算法在不增加噪声功能比例的前提下能够注释更多的蛋白质.PEFM算法的敏感性明显低于NC.这是因为PEFM算法只选择了排名靠前的部分功能用于注释功能未知的蛋白质,而NC方法是将邻居的所有功能全部赋予测试的蛋白质.但是这种策略导致NC方法预测的功能中包含大量的噪声功能,使得特异性急剧下降.本次实验中,虽然NC方法的敏感性比PEFM提高了12.93%,但是特异性却比PEFM下降了236.3%.因此,就综合性能而言,PEFM方法的F-measure值分别比D-PIN,FPM,Zhang, DCS,NC和PON提高1.71%,20.72%,90.43%, 35.28%,114.53%和192.33%.由此可见,PEFM方法具有最高综合性能.
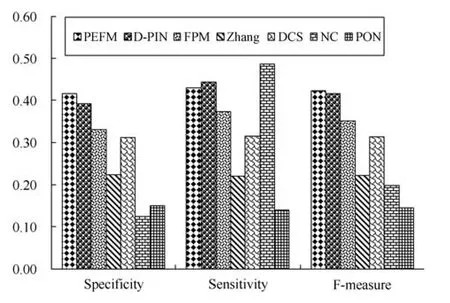
图7 各种算法综合性能对比Fig.7 Overall performance comparison of various algorithms
为了更加全面、客观地对比分析各种方法的性能,我们将尽可能地为各种方法选择相同的功能数量选取策略,对每一个蛋白质,分别选取各种方法预测的前K项功能进行预测.针对Zhang方法和DCS方法,选取前M(M≤K)个最相似的蛋白质,从这M个蛋白质的功能列表中选取前K项功能作为预测的功能.功能根据蛋白质的相似值的最大值降序排列(例如,有多个蛋白质具有某项功能Fi,则取这些蛋白质中与待预测的蛋白质最相似的蛋白质的相似值作为功能Fi的排序得分);对于D-PIN, FPM,PEFM,NC和PON方法,我们分别选取各自方法预测的前K个GO Term对功能未知的蛋白质进行功能注释.K的取值从1∼50,对于不同的K值,分别计算各种方法的平均F-measure值,对比结果如图8所示.
图8清晰地显示,当K从1增长到50时, PEFM始终具有最高的平均F-measure值.随着K值的增长,PEFM方法的F-Measure值虽然略微有所波动,但基本能维持在0.33左右,Zhang方法和DCS方法的F-Measure值则下降非常明显,这说明K的选取对于PEFM算法的影响不大.
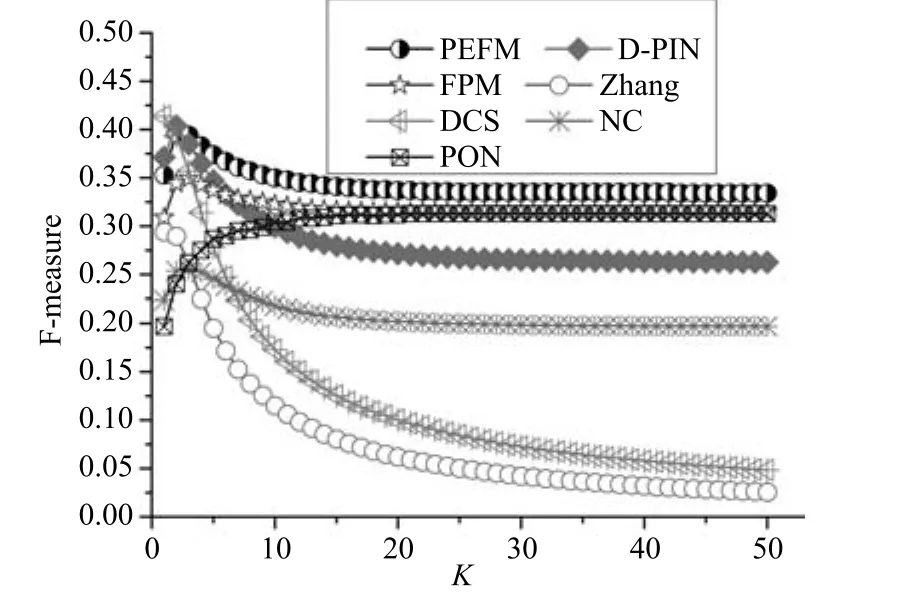
图8 不同K值时各种算法的F-measure对比Fig.8 Comparison of average F measure of various algorithms under diあerent K values
2.5 留部分法验证
我们已经采用留一法测试了PEFM算法的性能,实验结果表明,PEFM方法确实在现有方法的基础上提高了预测准确率.实际应用中,很多蛋白质的功能是缺失的.本节将采用留部分法测试PEFM方法是否能在部分蛋白质功能缺失的情形下依然保持较高的准确率.图9是留部分法实验结果.
我们随机移除10%、20%、50%和80%蛋白质的功能信息,这部分蛋白质作为测试集,剩余蛋白质为训练集,用于对这部分蛋白质进行功能注释.为尽量降低随机性对实验结果造成的误差,我们对每个方法运行1000次,取平均值作最终结果.
从图9不难发现,即便是移除10%,20%,50%和80%的蛋白质后,PEFM方法依然获得最高的F-Measure值,且优势比较明显.即便部分蛋白质的功能信息缺失,该方法依然能够取得优于现有功能预测方法的性能.
2.6 其他数据集结果
为了全面对比各种功能预测算法,我们还采用留一法在其他三个不同的酵母相互作用网络(Krogan数据集、Gavin数据集和BioGrid数据集)测试了PEFM方法和其他六种对比方法.表2列出了不同方法在三个网络上预测功能的实验结果.

图9 留部分法实验结果Fig.9 Results of leave-percent-out cross validation
从表2可以看出,采用留一法在三个网络进行功能预测时,PEFM依然取得最高特异性和F-measure值.在不同数据集上的测试结果也证明了特异性算法的有效性.综合上述分析,相比其他几种功能预测算法,PEFM算法具有最高的预测准确率.
3 结论
现有的蛋白质功能预测方法整合PPI网络和多元生物信息数据,从而提高功能预测性能.而融入多元信息后,蛋白质之间的相互作用变得多样化.不同类型的相互作用在功能预测中的作用各不相同.将两个蛋白质间的多种相互作用进行简单合并,虽然能有效地降低假阴性的影响,增加预测的功能数量,但同时也增加了假阳性功能的数量,使得功能预测的整体性能提高不大.本文利用网络拓扑特性、结构域信息和复合物信息构造多关系的蛋白质相互作用网络.鉴于蛋白质功能与模块之间的紧密联系,本文从多关系网络中挖掘关键功能模块,利用关键功能模块的功能对蛋白质进行功能注释.四个酵母的PPI网络上的实验结果验证了方法的有效性.
1 Zhao B H,Wang J X,Li M,Li X Y,Li Y H,Wu F X,Pan Y.A new method for predicting protein functions from dynamic weighted interactome networks.IEEE Transactions on NanoBioscience,2016,15(2):131−139
2 Schwikowski B,Uetz P,Fields S.A network of proteinprotein interactions in yeast.Nature Biotechnology,2000, 18(12):1257−1261
3 Dutkowski J,Ideker T.Protein networks as logic functions in development and cancer.PLoS Computational Biology, 2011,7(9):e1002180
4 Hu Sai,Xiong Hui-Jun,Zhao Bi-Hai,Li Xue-Yong,Wang Jing.Construction of dynamic-weighted protein interactome network and its application.Acta Automatica Sinica,2015, 41(11):1893−1900
(胡赛,熊慧军,赵碧海,李学勇,王晶.动态加权蛋白质相互作用网络构建及其应用研究.自动化学报,2015,41(11):1893−1900)
5 Zhao B H,Wang J X,Li X Y,Wu F X.Essential protein discovery based on a combination of modularity and conservatism.Methods,2016,110:54−63
6 Li X Y,Wang J X,Zhao B H,Wu F X,Pan Y.Identi fication of protein complexes from multi-relationship protein interaction networks.Human Genomics,2016,10(S2):17
7 Hu Sai,Xiong Hui-Jun,Li Xue-Yong,Zhao Bi-Hai,Ni Wen-Yin,Yang Pin-Hong,Liu Zhen.Construction of multirelation protein networks and its application.Acta Automatica Sinica,2015,41(12):2155−2163
(胡赛,熊慧军,李学勇,赵碧海,倪问尹,杨品红,刘臻.多关系蛋白质网络构建及其应用研究.自动化学报,2015,41(12):2155−2163)
8 Peng W,Wang J X,Cai J,Chen L,Li M,Wu F X.Improving protein function prediction using domain and protein complexes in PPI networks.BMC Systems Biology,2014, 8(1):35
9 Zotenko E,Mestre J,O′Leary D P,Przytycka T M.Why do hubs in the yeast protein interaction network tend to be essential:reexamining the connection between the network topology and essentiality.PLoS Computational Biology,2008,4(8):e1000140
10 Nepusz T,Yu H Y,Paccanaro A.Detecting overlapping protein complexes in protein-protein interaction networks.Nature Methods,2012,9(5):471−472
11 Xenarios I,Rice D W,Salwinski L,Baron M K,Marcotte E M,Eisenberg D.DIP:the database of interacting proteins.Nucleic Acids Research,2000,28(1):289−291
12 Stark C,Breitkreutz B J,Chatr-Aryamontri A,Boucher L, Oughtred R,Livstone M S,Nixon J,Van Auken K,Wang X D,Shi X Q,Reguly T,Rust J M,Winter A,Dolinski K, Tyers M.The BioGRID interaction database:2011 update.Nucleic Acids Research,2011,39(S1):D698−D704
13 Gavin A C,Aloy P,Grandi P,Krause R,Boesche M, Marzioch M,Rau C,Jensen L J,Bastuck S,D¨umpelfeld B, Edelmann A,Heurtier M A,Hoあman V,Hoefert C,Klein K,Hudak M,Michon A M,Schelder M,Schirle M,Remor M,Rudi T,Hooper S,Bauer A,Bouwmeester T,Casari G, Drewes G,Neubauer G,Rick J M,Kuster B,Bork P,Russell R B,Superti-Furga G.Proteome survey reveals modularity of the yeast cell machinery.Nature,2006,440(7084):631−636
14 Krogan N J,Cagney G,Yu H Y,Zhong G Q,Guo X H, Ignatchenko A,Li J,Pu S Y,Datta N,Tikuisis A P,Punna T,Peregr´ın-Alvarez J M,Shales M,Zhang X,Davey M, Robinson M D,Paccanaro A,Bray J E,Sheung A,Beattie B,Richards D P,Canadien V,Lalev A,Mena F,Wong P,Starostine A,Canete M M,Vlasblom J,Wu S,Orsi C, Collins S R,Chandran S,Haw R,Rilstone J J,Gandi K, Thompson N J,Musso G,Onge P S,Ghanny S,Lam M H Y,Butland G,Altaf-Ul A M,Kanaya S,Shilatifard A, O′Shea E,Weissman J S,Ingles C J,Hughes T R,Parkinson J,Gerstein M,Wodak S J,Emili A,Greenblatt J F. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae.Nature,2006,440(7084):637−643
15 Martin D M A,Berriman M,Barton G J.GOtcha:a new method for prediction of protein function assessed by the annotation of seven genomes.BMC Bioinformatics,2004, 5(1):178
16 Lima T,Auchincloss A H,Coudert E,Keller G,Michoud K,Rivoire C,Bulliard V,de Castro E,Lachaize C,Baratin D,Phan I,Bougueleret L,Bairoch A.HAMAP:a database of completely sequenced microbial proteome sets and manually curated microbial protein families in UniProtKB/Swiss-Prot.Nucleic Acids Research,2009,37(S1):D471−D478
17 Hawkins T,Chitale M,Luban S,Kihara D.PFP:automated prediction of gene ontology functional annotations with confidence scores using protein sequence data.Proteins:Struc-ture,Function,and Bioinformatics,2009,74(3):566−582
18 Pu S Y,Wong J,Turner B,Cho E,Wodak S J.Up-todate catalogues of yeast protein complexes.Nucleic Acids Research,2009,37(3):D825−D831
19 Zhang S,Chen H,Liu K,Sun Z R.Inferring protein function by domain context similarities in protein-protein interaction networks.BMC Bioinformatics,2009,10(1):395
20 Liang S D,Zheng D D,Standley D M,Guo H R,Zhang C. A novel function prediction approach using protein overlap networks.BMC Systems Biology,2013,7(1):61
