定向进化技术的最新进展
2018-02-28曲戈赵晶郑平孙际宾孙周通
曲戈,赵晶,郑平,孙际宾,孙周通
1 中国科学院天津工业生物技术研究所,天津 300308
2 工业酶国家工程实验室,天津 300308
3 中国科学院系统微生物工程重点实验室,天津 300308
自20世纪后半叶以来,随着分子生物学和蛋白分离纯化技术的进步,蛋白质在生物技术、工业生产、医药食品、基因治疗和环境保护等领域扮演了越来越重要的角色[1-2]。但作为生物催化剂,天然酶存在诸如立体/区域选择性差、底物谱窄、催化效率低、稳定性差及产物抑制性等问题[3-4],严重阻碍了生物催化剂的广泛应用。
为加快对生物催化剂——酶已有性能的改造提升及新功能开发应用,近20年来涌现了一系列蛋白质定向进化技术,如饱和突变、易错PCR和DNA混组等。通过在实验室条件下模拟自然进化过程,从构建的突变体文库中筛选到能满足特定需求的目标突变体[3-4],大大拓展了酶的应用范围。如改造后的氧化酶可直接使用氧分子作为电子受体,实现二氧化碳的高效固定[5];CRISPR-Cas9蛋白可识别的序列范围也更大、更精准[6];重组酶可高效识别HIV病毒基因两侧特异性位点并将其从宿主基因组中切除[7];能催化卡宾反应的仿生金属酶[8]和实现转氨酶法合成西他列汀的工业化应用[9],都得益于定向进化技术的发展应用。当今的定向进化技术整合了有理设计、适度的随机突变和高效筛选,可在已知或未知目标蛋白质结构信息及催化机制的情况下,对蛋白质进行针对性改造。然而,蛋白定向进化技术在上述领域中的应用潜力还远没有被挖掘,其主要挑战在于如何设计构建高质量的多样性突变体文库和建立高效、快速的筛选方法[10-11]。本文主要从突变体文库的构建方面综述蛋白质定向进化领域涌现的新方法及其应用,并对发展前景进行展望。
1 定向进化策略
根据突变体文库构建方法的不同,定向进化可分为非理性设计、半理性设计和理性设计3种策略。其大致思路是通过模拟自然进化,对目的基因进行重复多轮的突变、表达和筛选,从而在短时间内完成自然界中需要成千上万年的进化,最终获得性能改进或具有新功能的蛋白质[12]。
1.1 非理性设计 (Non-rational design)
非理性设计即随机进化策略,优点是不需要对酶序列及结构有深入了解,仅需通过随机突变和片段重组的方法模拟自然进化。1978年,Michael Smith首次提出定点突变技术(Site-specific mutagenesis),开启了蛋白质改造与设计的大门[13]。自此以后,一系列经典的基因突变方法被开发应用,主要包括饱和突变(Saturation mutagenesis,SM)、 易 错 PCR(Error-prone polymerase chain reaction,epPCR) 及DNA重组 (DNA shuffling)。
epPCR概念由Leung团队于1989年提出[14],然而首次将 epPCR应用于酶改造却是 3年后由Hawkins等进行的体外抗体筛选[15]。其基本原理是通过改变PCR反应体系的反应条件或使用低保真的 DNA聚合酶,增加碱基随机错配率,从而造成多点突变,产生序列多样性的突变体文库,因其不需要蛋白结构信息、操作简单而被研究者广泛采用。然而该技术的应用受到以下几方面制约:聚合酶的碱基偏好性 (通常 AG>TC)、突变效率低且缺少后续突变 (每轮每基因仅 3–5个突变) 等[16]。Arnold团队于1993年开创性地使用多轮epPCR (sequential epPCR),连续反复地对枯草杆菌蛋白酶进行随机突变,逐步提高了突变体在有机溶剂 DMF中的稳定性[17]。通常情况下需要至少连续4轮的epPCR逐步积累正向突变,才能获得酶性能显著提高的目的突变体。
1994年,Stemmer团队开发了DNA shuffling技术,主要用于单基因或多基因的重组,不仅可加速有义突变的积累,还能组合两个或多个已优化的参数,并成功提高了β-内酰胺酶的活性[18]。该技术利用DNase将一组带有有义突变位点的同源基因切成随机片段 (通常10−50 bp),使用PCR使之延伸重组获得全长基因。优点是操作简单,不需要蛋白结构信息,容易获得有义突变;缺点是要求基因序列间至少具有70%的一致性。
20世纪80年代Wells团队提出寡核苷酸饱和突变(Oligonucleotide-based saturation mutagenesis,OSM)[19],可实现单点饱和突变。Reetz等利用寡聚核苷酸重组技术实现了多位点饱和突变(Assembly of designed oligonucleotides,ADO)[20]。接着Schwaneberg团队开发了序列饱和突变技术(Sequence saturation mutagenesis,SeSaM)[21],能较好地克服 DNA聚合酶的碱基偏好性并提高突变效率;但因其操作繁琐、试验周期长而较少被采用[16]。此外,饱和突变技术还可与epPCR、DNA shuffling等技术组合使用,迅速积累有义突变,得到最佳突变组合的酶基因。如 Reetz团队率先综合利用这3项传统技术,成功提高了脂肪酶的对映体选择性[22-23]。
1.2 理性设计 (Rational design)
理性设计是一种智能改造手段,依赖计算机技术 (in silico) 模拟自然界蛋白质的进化轨迹,通过计算机虚拟突变,筛选可快速准确预测目标突变体。通过一系列基于生物信息学开发的算法和程序[24],预测蛋白质活性位点并考察特定位点突变对其稳定性、折叠及与底物结合等方面的影响,从而对蛋白质进行针对性地改造和模拟筛选[25]。
在当前第3次酶改造浪潮中,基于计算机辅助设计和大尺度的分子动力学模拟可高效、快捷地改造和筛选生物催化剂[26-27],不仅可高精度地预测蛋白结构[28],还可从头设计自然界中不存在的新酶[29],以及对现有酶进行改造,赋予其新功能。如改造后的细胞色素 C氧化酶可提高碳–硅键形成的催化效率[30];赋予 30亿年来逆转录酶原本不存在的校对功能[31];从头设计能催化Kemp 消除反应的新酶[32],及新的(β/α)8TIM 桶蛋白[33]等。
尽管新酶设计已取得一定成功,但依然面临诸多挑战:首先,其成功率较低;其次,计算工作繁重,对计算机资源依赖非常高;再次,设计出的新酶结构和稳定性较差,催化活性往往偏低。主要是因为对酶序列/结构/功能之间关系的认识还不够深入。
1.3 半理性设计 (Semi-rational design)
半理性设计主要借助生物信息学方法,基于同源蛋白序列比对、三维结构或已有知识,理性选取多个氨基酸残基作为改造靶点,结合有效密码子的理性选用,通过构建高质量突变体文库,有针对性地对蛋白质进行改造[10]。常见的半理性策略如表1所示。

表1 半理性设计常用策略Table 1 Strategies of semi-rational design
近年来,结合非理性和理性设计的半理性设计,兼顾了序列空间多样性 (Sequence space) 和筛选工作量,是一种应用非常广泛的定向进化技术[44-45]。通过Web of Science 数据库检索主题“相应技术名称” AND “biocatalysis”,自 2005 年至今(截止2017年4月25日),生物催化领域应用DNA shuffling和 epPCR的文献数目共计57篇,应用从头设计 (de novodesign) 的有62篇,而使用SM的有140篇 (图1)。由此可见,生物催化领域中半理性设计策略占据了主导地位。
1.3.1 组合活性中心饱和突变和迭代饱和突变
通过对来自铜绿假单胞菌Pseudomonas aeruginosa的脂肪酶及其他酶改造发现,对手性选择有义的突变主要集中在酶催化口袋区域。Reetz团队由此提出了组合活性中心饱和突变策略 (Combinatorial active-site saturation test,CAST)[43]:基于序列和/或结构信息,借助计算机模拟在酶催化活性中心周围选取与底物有直接相互作用的氨基酸残基,通过理性分组进行单轮或多轮迭代饱和突变 (Iterative saturation mutagenesis,ISM)。一般情况下单轮突变难以达到预期目标,需要进行多轮叠加突变。为减少筛选规模,通常将2−4个氨基酸残基分为一组。这些残基在空间上彼此靠近,往往具有协同作用。Kazlauskas团队统计分析并比较了位于活性中心和远离活性中心的氨基酸位点突变对酶的活性、底物选择性、对映体选择性及稳定性的影响,指出前者更有利于改造酶的底物选择性及对映体选择性,从统计学的角度进一步支持了CAST方法的可靠性[46]。因此CAST方法广泛应用于酶的立体/区域选择性、底物谱、催化效率等参数的改造[47-48]。

图1 定向进化三大策略(无理、半理性和理性设计)在生物催化领域的文献发表(数据来自Web of Science,采用主题查询方式,自2005年至2017年4月25日)Fig. 1 The number of publications on directed evolution (non-, semi- and rational design) in the field of biocatalysis. Data were collected from Web of Science since 2005 to April 25, 2017, using Topic search. DS:DNA shuffling; epPCR: error-prone PCR; SM: saturation sutagenesis; de novo:de novo design. “+” represents AND biocatalysis; Dashed line denotes the trend of publication numbers.
以4位点 (A、B、C和D) 的ISM系统为例(图 2A),一共有 24条进化路径,其中每个位点可包括1至多个氨基酸残基,对每个位点分别进行饱和突变,产生4个不同的突变体文库。在进行下一轮迭代突变时,已进行过突变的位点保留。因此理论上4个位点全部完成迭代突变共需4轮,即构建64个突变体文库。然而实际上由于仅选取每轮迭代筛选到的最优突变体进行下一轮突变,所以构建的突变体文库数目远小于理论值。为了防止进入进化路线中局部最优的“死胡同” (红色虚线,图 2B),可从上轮筛选中选取性能次好的突变体作为模板,进行后续的迭代突变[4,49]。
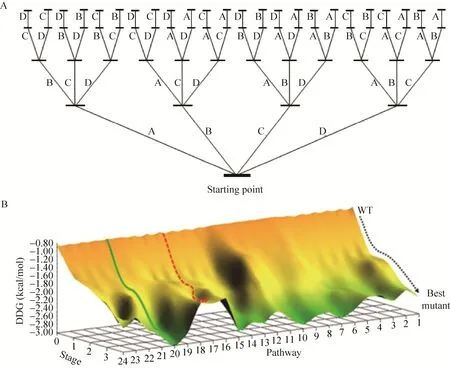
图2 ISM系统进化路径与能垒分布[4]Fig. 2 Representation of ISM system featuring all of the evolutionary pathways and energy barrier distributions[4]. (A)ISM scheme for 4-site system involving 24 upward pathways. (B) Experimental fitness-pathway landscape featuring all 24 pathways of a four-site ISM system using an epoxide hydrolase as catalyst in the kinetic resolution of a racemic epoxide. Green line: typical trajectory in which every mutant library in the four-step sequence contains an improved mutant (no local minima). Red line: typical trajectory in which at least one library along the four-step sequence is devoid of an improved mutant (local minimum).
2 单密码子和三密码子饱和突变
筛选工作是酶定向改造中最重要的瓶颈[26,48]。基于CAST/ISM的半理性设计方法试图通过建立针对特定改造靶点的高质量突变体文库来解决筛选问题[50]。虽然获得了极大成功,然而随着拟突变位点数目的增加,筛选规模呈指数级增长。以NNK简并密码子为例,设定95%文库覆盖度,如果同时突变10个位点时,需筛选3.4×1015个转化子,即使采用 NDT为简并密码子,仍需筛选1.9×1011个转化子 (表2)。因此需要设计开发更加高效的建库策略来解决酶定向进化的筛选问题。
为降低筛选规模,仅使用单密码子对酶催化口袋进行扫描,称之为单密码子饱和突变(SCSM)[51-52]。基于酶催化口袋的理化性质 (如亲疏水性)以及已有信息,理性选取某一特定的氨基酸密码子作为建构单元,重塑酶催化口袋,达到提高或反转立体选择性目的[52]。以同时突变 10个位点为例,在95%覆盖度下,筛选规模从NNK的约3×1015或NDT的2×1011(表2),降至约3 000。该方法的难点在于单密码子的选择,并缺少多密码子间的协同作用。
为进一步降低筛选工作量,基于蛋白序列(多重序列同源比对确定保守位点) 及结构 (晶体结构或同源建模) 的相关信息,结合酶的催化性质及已知实验数据支持,理性选择3种氨基酸密码子作为饱和突变的建构单元,然后将拟突变的多个位点进行理性分组 (3−4个氨基酸残基分为一组),该策略称之为三密码子饱和突变 (TCSM)。仍以同时突变10个位点为例,突变体文库容量约为 3.14×106;而如果将该 10位点分为 3组(4+3+3),并分别构建突变体文库,那么突变体文库的总体规模则可降至1 152 (768+192+192)。由此可见,选取合适的简并密码子,将多个拟突变位点理性分为若干组并分别构建突变体文库,结合ISM策略可有效减少转化子筛选量[53-54]。
综上所述,定向进化技术从诞生发展至今,经历了从随机到半理性的过程,同时借助计算机技术向理性方向发展。在此过程中依据基础和应用研究的需要,涌现了一系列实用的新技术,主要是为了平衡因不同建库策略所带来的序列多样性和筛选工作量问题,重在提高文库构建的质量。定向进化发展过程中的重要事件总结如图 3所示,未来走向基于计算机技术的理性设计是必然,但任重而道远,半理性设计依然会成为未来10年内的主流技术。
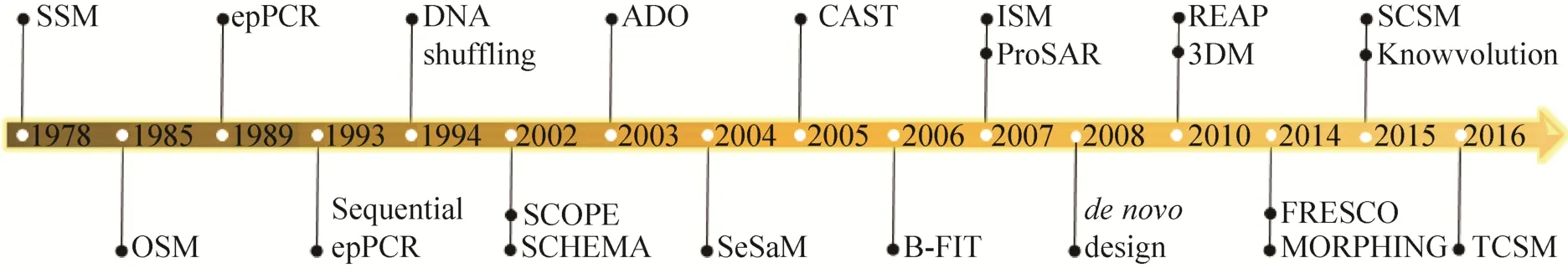
图3 定向进化技术大事记Fig. 3 Events in directed evolution.
3 定向进化新技术在立体选择性等方面的应用
以柠檬烯环氧水解酶 (LEH) 催化环氧己烷合成手性环己烷-1,2-二醇为模式反应。借助计算机模拟选取 LEH疏水口袋与底物直接相互作用的10个氨基酸残基(L74/F75/M78/I80/L103/L114/I116/F134/F139/L147)作为改造靶点,理性选取疏水的缬氨酸 (V) 和苯丙氨酸 (F) 分别作为构建单元,对所选取的 10个位点进行SCSM组合扫描及迭代饱和突变。总筛选约4 000个转化子,成功获得了立体选择性提高和反转的突变体,将ee值从野生型的2% (S,S)-构型提高到了92%,而(R,R)-构型的达到 96%[52]。为进一步降低筛选规模,提高建库质量,重新选取 10个氨基酸残基(M32/L35/L74/M78/I80/V83/L103/L114/I116/L147)作为改造靶点,随机分为3组(A: I80/V83/L114/I116;B: L74/M78/L147; C: M32/L35/L103) (图 4)。理性选取 V-F-Y为氨基酸密码子作为构建单元,进行TCSM建库和迭代饱和突变。在库A中直接获得了 (S,S)-构型ee值提高至99%突变体和反转突变(R,R)-构型达到 89%,通过 1−2步 TCSM迭代饱和突变分别访问B库和/或C库,(R,R)-构型的ee值可达到97%,总筛选规模约1 000个转化子[53]。同样以LEH为例,采用TCSM方法同时对立体选择性、热稳定性和催化活力进行共进化,通过TCSM 设计和建库,虽然催化效率略有降低,但成功将ee值提高至94% (S,S)和80% (R,R),同时热稳定性提高了5−10 ℃,首次实现了立体选择性和热稳定性的共进化[55]。
为进一步验证TCSM的通用性,以嗜热厌氧菌Thermoanaerobacter brockii来源的醇脱氢酶(TbSADH) 不对称催化 α,α′碳骨架非常近似的羰基还原为手性醇。尝试化学法难以催化的反应,如四氢呋喃-3-酮、四氢噻吩-3-酮还原为手性醇(分别用于合成抗艾滋病药物安普那韦、膦沙那韦和第三代抗生素硫培南类药物)。野生型TbSADH对四氢呋喃-3-酮的立体选择性仅有23%且偏向于R-构型。通过TCSM设计和建库,分别理性选取缬氨酸-天冬酰胺-亮氨酸 (V-N-L) 和缬氨酸-谷氨酰胺-亮氨酸 (V-Q-L) 作为R-和S-构型改造的构建单元,将5个拟突变位点理性分为两组,用来构建和筛选R-和S-选择性的突变体文库,分别筛选576个转化子后即获得了系列提高的R-和S-选择性突变体,ee值达到95%−99%[54]。
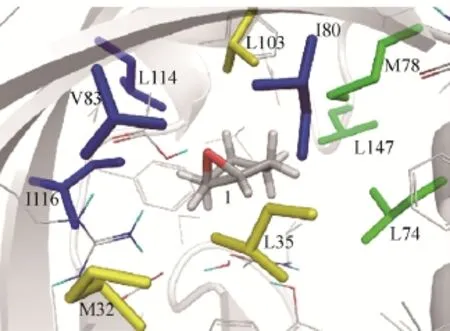
图4 LEH结合口袋10个位点的选取及随机分组Fig. 4 Ten residues lining the binding pocket of LEH,assigned to three randomization sites: A (blue), B (green),and C (yellow). Substrate 1 is shown by grey sticks.
TCSM的另一个应用实例是对来自巨大芽孢杆菌自给型细胞色素 P450-BM3单加氧酶的定向进化改造。基于野生型 P450-BM3的晶体结构及前期对酶结合口袋的研究,通过理性选择突变位点及构建密码子单元,Li等成功地改造了该酶的区域选择性、立体选择性和底物适配性,并结合醇脱氢酶 (ADH) 设计级联反应,以惰性石油基化合物环己烷为原料,可按设计一锅法分别合成相应 (R,R)-型、(S,S)-型或消旋型手性二醇产物[56]。
因此,SCSM和TCSM策略的设计可大幅提高建库质量和降低筛选规模,在保证95%文库覆盖度的情况下,与使用 NNK密码子相比,筛选工作量降低了 1012倍。并加速对酶的立体选择/区域选择性改造,已成功应用于对柠檬烯环氧水解酶 (LEH)[53,55]、醇脱氢酶 (ADH)[54,57]和细胞色素P450单加氧酶[56]的改造。
4 结语及展望
近年来定向进化技术广泛应用于生物催化剂改造,但筛选工作是限制酶定向进化改造的瓶颈。探索高质量突变体文库的构建及筛选新方法是定向进化领域的重要研究内容。基于CAST/ISM的原理,孙周通等开发了系列快速半理性设计方法,如SCSM和TCSM,通过建立“小而精”的高质量突变体文库来解决筛选问题,为有机化学和生物技术中酶的改造和应用提供了新策略。
TCSM 解决了定向进化一直存在的筛选瓶颈,总体筛选规模可控制在1 000左右,单文库可控制在500左右,甚至更低 (<200)。使定向进化不再依赖于筛选方法,可应用于酶的立体选择性、区域选择性及底物适配性等多参数并行改造,快速高效设计生物催化剂应用于药物前体催化,解决化学法难以催化的反应等。不难想象,TCSM将会受到有机催化、生物催化、代谢工程与合成生物技术等领域研究人员更多的关注与应用。此外,作者也尝试了利用双密码子饱和突变(DCSM)[58],即采用两个密码子作为构建单元重塑酶催化口袋。同样以 LEH的立体选择性改造为例,与SCSM和TCSM进行了系统的比较研究,拓展了定向进化技术工具箱[58]。然而无论哪种密码子饱和突变策略,均依赖酶的保守位点、三维结构和/或已有知识,可根据改造对象和应用目的选择合适突变策略及对其进行组合应用。
除了构建“小而精”的突变体文库可有效克服筛选瓶颈外,将突变体筛选工作转移至计算机进行虚拟筛选,也是快速获得目标突变体的有效方式。2015年,Dick Janssen和David Baker团队合作,将LEH催化中心附近的11个位点突变为疏水性残基,并利用分子动力学模拟等计算机技术对突变体文库进行虚拟筛选,最终仅通过实验构建验证 37个突变体即获得成功[59]。组合使用基于计算机设计大规模初筛和实验室小规模二次筛选将成为解决筛选瓶颈的重要策略,同时随着基因合成成本的降低,结合突变体文库的全合成建库策略,可以有效减少密码子引入的偏好性和提高文库序列多样性,将成为定向进化技术重要的发展方向。
[1]Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol, 2009, 10(12): 866–876.
[2]Packer MS, Liu DR. Methods for the directed evolution of proteins. Nat Rev Genet, 2015, 16(7):379–394.
[3]Bommarius AS. Biocatalysis: a status report. Annu Rev Chem Biomol Eng, 2015, 6(1): 319–345.
[4]Reetz MT. Biocatalysis in organic chemistry and biotechnology: past, present, and future. J Am Chem Soc, 2013, 135(34): 12480–12496.
[5]Schwander T, Schada von Borzyskowski L, Burgener S, et al. A synthetic pathway for the fixation of carbon dioxidein vitro. Science, 2016, 354(6314):900–904.
[6]Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature, 2015, 523(7561): 481–485.
[7]Karpinski J, Hauber I, Chemnitz J, et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity.Nat Biotechnol, 2016, 34(4): 401–409.
[8]Dydio P, Key HM, Nazarenko A, et al. An artificial metalloenzyme with the kinetics of native enzymes.Science, 2016, 354(6308): 102–106.
[9]Savile CK, Janey JM, Mundorff EC, et al.Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture.Science, 2010, 329(5989): 305–309.
[10]Cheng F, Zhu LL, Schwaneberg U. Directed evolution 2.0: improving and deciphering enzyme properties. Chem Commun, 2015,51(48):9760–9772.
[11]Denard CA, Ren HQ, Zhao HM. Improving and repurposing biocatalysts via directed evolution. Curr Opin Chem Biol, 2015, 25: 55–64.
[12]Sheldon RA, Pereira PC. Biocatalysis engineering:the big picture. Chem Soc Rev, 2017, 46(10):2678–2691.
[13]Hutchison III CA, Phillips S, Edgell MH, et al.Mutagenesis at a specific position in a DNA sequence. J Biol Chem, 1978, 253(18): 6551–6560.
[14]Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction.Technique, 1989, 1: 11–15.
[15]Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity: mimicking affinity maturation. J Mol Biol, 1992, 226(3):889–896.
[16]Ruff AJ, Dennig A, Schwaneberg U. To get what we aim for——Progress in diversity generation methods.FEBS J, 2013, 280(13): 2961–2978.
[17]Chen KQ, Arnold FH. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci USA, 1993,90(12): 5618–5622.
[18]Stemmer WPC. Rapid evolution of a protein in vitro by DNA shuffling. Nature, 1994, 370(6488):389–391.
[19]Wells JA, Vasser M, Powers DB. Cassette mutagenesis: an efficient method for generation of multiple mutations at defined sites. Gene, 1985,34(2/3): 315–323.
[20]Zha DX, Eipper A, Reetz MT. Assembly of designed oligonucleotides as an efficient method for gene recombination: a new tool in directed evolution.ChemBioChem, 2003, 4(1): 34–39.
[21]Wong TS, Tee KL, Hauer B, et al. Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution. Nucleic Acids Res, 2004, 32(3):e26.
[22]Reetz MT, Zonta A, Schimossek K, et al. Creation of enantioselective biocatalysts for organic chemistry by in vitro evolution. Angew Chem Int Ed, 1997, 36(24):2830–2832.
[23]Reetz MT, Wilensek S, Zha DX, et al. Directed evolution of an enantioselective enzyme through combinatorial multiple-cassette mutagenesis. Angew Chem Int Ed, 2001, 40(19): 3589–3591.
[24]Ebert MC, Pelletier JN. Computational tools for enzyme improvement: why everyone can- and should-use them. Curr Opin Chem Biol, 2017, 37:89–96.
[25]Huang PS, Boyken SE, Baker D. The coming of age ofde novoprotein design. Nature, 2016, 537(7620):320–327.
[26]Bornscheuer UT, Huisman GW, Kazlauskas RJ, et al.Engineering the third wave of Biocatalysis. Nature,2012, 485(7397): 185–194.
[27]Privett HK, Kiss G, Lee TM, et al. Iterative approach to computational enzyme design. Proc Natl Acad Sci USA, 2012, 109(10): 3790–3795.
[28]Ovchinnikov S, Park H, Varghese N, et al. Protein structure determination using metagenome sequence data. Science, 2017, 355(6322): 294–298.
[29]Lin YR, Koga N, Tatsumi-Koga R, et al. Control over overall shape and size in de novo designed proteins.Proc Natl Acad Sci USA, 2015, 112(40):E5478–E5485.
[30]Kan SBJ, Lewis RD, Chen K, et al. Directed evolution of cytochrome c for carbon-silicon bond formation: bringing silicon to life. Science, 2016,354(6315): 1048–1051.
[31]Ellefson JW, Gollihar J, Shroff R, et al. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science, 2016, 352(6293): 1590–1593.
[32]Röthlisberger D, Khersonsky O, Wollacott AM, et al.Kemp elimination catalysts by computational enzyme design. Nature, 2008, 453(7192): 190–195.
[33]Huang PS, Feldmeier K, Parmeggiani F, et al.De novodesign of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat Chem Biol,2015, 12(1): 29–34.
[34]O'Maille PE, Bakhtina M, Tsai MD. Structure-based combinatorial protein engineering (SCOPE). J Mol Biol, 2002, 321(4): 677–691.
[35]Wijma HJ, Floor RJ, Jekel PA, et al. Computationally designed libraries for rapid enzyme stabilization.Protein Eng Des Sel, 2014, 27(2): 49–58.
[36]Chen F, Gaucher EA, Leal NA, et al. Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc Natl Acad Sci USA, 2010,107(5): 1948–1953.
[37]Kuipers RK, Joosten HJ, van Berkel WJH, et al.3DM: systematic analysis of heterogeneous superfamily data to discover protein functionalities.Proteins, 2010, 78(9): 2101–2113.
[38]Fox RJ, Davis SC, Mundorff EC, et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat Biotechnol, 2007, 25(3): 338–344.
[39]Gonzalez-Perez D, Molina-Espeja P, Garcia-Ruiz E,et al. Mutagenic organized recombination process by homologousin vivogrouping (MORPHING) for directed enzyme evolution. PLoS ONE, 2014, 9(3):e90919.
[40]Gutierrez EA, Mundhada H, Meier T, et al.Reengineered glucose oxidase for amperometric glucose determination in diabetes analytics. Biosens Bioelectron, 2013, 50(4): 84–90.
[41]Voigt CA, Martinez C, Wang ZG, et al. Protein building blocks preserved by recombination. Nat Struct Mol Biol, 2002, 9(7): 553–558.
[42]Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability.Angew Chem Int Ed, 2006, 45(46): 7745–7751.
[43]Reetz MT, Bocola M, Carballeira JD, et al.Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test.Angew Chem Int Ed, 2005, 44(27): 4192–4196.
[44]Lutz S. Beyond directed evolution-semi-rational protein engineering and design. Curr Opin Biotechnol, 2010, 21(6): 734–743.
[45]Chica RA, Doucet N, Pelletier JN. Semi-rational approaches to engineering enzyme activity:combining the benefits of directed evolution and rational design. Curr Opin Biotechnol, 2005, 16(4):378–384.
[46]Morley KL, Kazlauskas RJ. Improving enzyme properties: when are closer mutations better? Trends Biotechnol, 2005, 23(5): 231–237.
[47]Reetz MT, Prasad S, Carballeira JD, et al. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc, 2010, 132(26): 9144–9152.
[48]Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution.ChemBioChem, 2008, 9(11): 1797–1804.
[49]Gumulya Y, Sanchis J, Reetz MT. Many pathways in laboratory evolution can lead to improved enzymes:how to escape from local minima. ChemBioChem,2012, 13(7): 1060–1066.
[50]Reetz MT, Sanchis J. Constructing and analyzing the fitness landscape of an experimental evolutionary process. ChemBioChem, 2008, 9(14):2260–2267.
[51]Sun ZT, Wikmark Y, Bäckvall JE, et al. New concepts for increasing the efficiency in directed evolution of stereoselective enzymes. Chem Eur J,2016, 22(15): 5046–5054.
[52]Sun ZT, Lonsdale R, Kong XD, et al. Reshaping an enzyme binding pocket for enhanced and inverted stereoselectivity: use of smallest amino acid alphabets in directed evolution. Angew Chem Int Ed,2015, 54(42): 12410–12415.
[53]Sun ZT, Lonsdale R, Wu L, et al. Structure-guided triple-code saturation mutagenesis: efficient tuning of the stereoselectivity of an epoxide hydrolase. ACS Catal, 2016, 6(3): 1590–1597.
[54]Sun ZT, Lonsdale R, Ilie A, et al. Catalytic asymmetric reduction of difficult-to-reduce ketones:triple-code saturation mutagenesis of an alcohol dehydrogenase. ACS Catal, 2016, 6(3): 1598–1605.
[55]Li GY, Zhang H, Sun ZT, et al. Multiparameter optimization in directed evolution: engineering thermostability, enantioselectivity, and activity of an epoxide hydrolase. ACS Catal, 2016, 6(6):3679–3687.
[56]Li AT, Ilie A, Sun ZT, et al. Whole-cell-catalyzed multiple regio- and stereoselective functionalizations in cascade reactions enabled by directed evolution.Angew Chem Int Ed, 2016, 55(39): 12026–12029.
[57]Sun ZT, Li GY, Ilie A, et al. Exploring the substrate scope of mutants derived from the robust alcohol dehydrogenase TbSADH. Tetrahedron Lett, 2016,57(32): 3648–3651.
[58]Sun ZT, Lonsdale R, Li GY, et al. Comparing different strategies in directed evolution of enzyme stereoselectivity: single- versus double-code saturation mutagenesis. ChemBioChem, 2016,17(19): 1865–1872.
[59]Wijma HJ, Floor RJ, Bjelic S, et al. Enantioselective enzymes by computational design andin silicoscreening. Angew Chem Int Ed, 2015, 54(12):3726–3730.
