Antibody drug conjugate development in gastrointestinal cancers: hopes and hurdles from clinical trials
2018-02-18XiaorongWuThomasKilpatrickIanChau
Xiaorong Wu, Thomas Kilpatrick, Ian Chau
Department of Medical oncology, Royal Marsden Hospital NHS foundation trust, Sutton SM2 5PT, UK.
Abstract Gastrointestinal (GI) cancers represent the leading cause of cancer-related mortality worldwide. Antibody drug conjugates (ADCs) are a rapidly growing new class of anti-cancer agents which may improve GI cancer patient survival.ADCs combine tumour-antigen specific antibodies with cytotoxic drugs to deliver tumour cell specific chemotherapy.Currently, only two ADCs [brentuximab vedotin and trastuzumab emtansine (T-DM1)] have been Food and Drug Administration approved for the treatment of lymphoma and metastatic breast cancer, respectively. Clinical research evaluating ADCs in GI cancers has shown limited success. In this review, we will retrace the relevant clinical trials investigating ADCs in GI cancers, especially ADCs targeting human epidermal growth receptor 2, mesothelin, guanylyl cyclase C, carcinogenic antigen-related cell adhesion molecule 5 (also known as CEACAM5) and other GI malignancy specific targets. We will review potential hurdles for their success and provide new perspective for future treatment.
Keywords: Antibody drug conjugates, human epidermal growth receptor 2, mesothelin, guanylyl cyclase C, carcinogenic antigen-related cell adhesion molecule 5 gastric cancer, colorectal cancer, pancreatic cancer, T-DM1, DS-8201a
INTRODUCTION
Gastrointestinal (GI) cancers comprise a heterogeneous group of cancers that include gastric, oesophageal,pancreatic, hepatobiliary, colorectal and anal cancers. GI cancers are the most common tumour type encountered worldwide and represent the leading cause of cancer-related mortality[1]. The only potentially curative strategy for GI cancers is surgery; however the majority of GI cancers are unresectable at diagnosis.Systemic or palliative treatments such as chemotherapy, radiotherapy, monoclonal antibody (mAb)therapy or combination therapies have greatly improved survival rates in the last decade; however, the 5 years survival of GI cancers remains poor. A new class of treatments under development is antibody drug conjugates (ADCs).
ADCs combine a tumour-antigen specific mAb with a cytotoxic drug (or “payload”) using a linker. ADCs exert their cytotoxic effects after the mAb directly binds to a tumour specific antigen and the ADC complex is internalized into the tumour cell. The complex is then degraded, which frees the cytotoxic drug to exert its cytotoxic effects on tumour cells. ADCs represent an attractive oncology treatment modality as they allow targeted delivery of cytotoxic agents directly to cancer cells, causing minimal toxicity to normal tissue[2-4].Only two ADCs are currently approved by US Food and Drug Administration and the European Medicines Agency. Brentuximab vedotin, targeting cancer antigen CD30, was first approved in 2011 for the treatment of patients with relapsed or refractory CD30-expressing Hodgkin's and anaplastic large cell lymphomas[5,6].T-DM1 (also known as trastuzumab emtansine or ado-trastuzumab emtansine) was approved in 2013 for use in patients with human epidermal growth receptor 2 (HER2)-expressing metastatic breast cancers who have disease progression following standard chemotherapy or HER2 targeted therapy[7]. Since 2013, more than 60 ADCs have reached clinical trials under investigation for haematological and oncological indications.
There are no ADCs in use for GI malignancies and many clinical trials that assess ADCs in GI cancers remain in the early stages. Due to the acting mechanism of ADCs, ideal target antigens are highly expressed on tumour cells and minimally expressed on healthy cells. GI potential tumour antigens under evaluation as ADC targets currently include HER2, mesothelin, guanylyl cyclase C (GCC), carcinogenic antigen-related cell adhesion molecule 5 (CEACAM5) and other tumour antigens. This review will examine ADC trials in GI cancers [Table 1], and discuss the possible challenges for their success and new strategies in the future to facilitate improvement of GI cancer management.
HER2 TARGETED ADCS
HER2 is a tyrosine protein kinase receptor which has been demonstrated to be expressed in certain GI malignancies, including gastro-oesophageal, gastric, pancreatic, gallbladder and colorectal cancers[8,9]. The HER2 mAb trastuzumab is the first biological therapy to have shown a median survival benefit of nearly 3 months in combination with chemotherapy for HER2 positive advanced gastric/gastro-oesophageal junction cancer compared to chemotherapy alone[10]. Thus, HER2 is a suitable target for ADCs in GI cancers.
T-DM1
Trastuzumab emtansine, or T-DM1, is made by conjugation of the microtubule polymerization inhibitor,emtansine, to trastuzumab via a noncleavable linker[11]. A phase I study of T-DM1 in breast cancer suggested that T-DM1 was well tolerated at maximum tolerated dose (MTD) of 2.4 mg/kg with potential antitumor activity[12]. However, despite significant efficacy and minimal toxicity in HER2 positive advanced breast cancer[13,14], T-DM1 was not superior to taxanes (docetaxel or paclitaxel) in patients with previously treated HER2 positive advanced gastric cancer[15]. The phase II/III GATSBY study enrolled patients with HER2-positive advanced gastric cancer who progressed during or after first-line therapy. Patients were randomly assigned to treatment groups of T-DM1 3.6 mg/kg every 3 weeks (n = 70) or 2.4 mg/kg weekly (n = 228)or a taxane (n = 117). The median overall survival (OS) was 7.9 months (95% CI 6.7-9.5) with T-DM1 2.4 mg/kg weekly and 8.6 months (95% CI 7.1-11.2) with taxane treatment (HR 1.15; 95% CI 0.87-1.51, onesided P = 0.86). The results of the secondary efficacy endpoints, including progression-free survival (PFS),objective response rate (ORR) and duration of response were consistent with the primary endpoints. In the GATSBY trial, the T-DM1 2.4 mg/kg group showed slightly lower incidence of ≥ grade 3 adverse events (AE)compared to taxane group. The most common ≥ grade 3 AE in the T-DM1 group were anaemia (26%) and thrombocytopenia (11%) compared with neutropaenia (39%) and anaemia (26%) in the taxane group[15]. As aconsequence, T-DM1 performed poorly as a second-line option for HER2 positive advanced gastric cancer.Several mechanisms may explain the absence of OS benefit in patients with advanced gastric cancer treated with T-DM1 compared to patients treated with taxanes. First, emtansine may have limited antitumor efficacy in gastric cancer; there is evidence showing that the microtubule polymerization inhibitor vinorelbine was less active in gastric cancer than in other cancers[16]. Additionally, patients in the GATSBY trial were selected based on archival HER2 status, which might be altered by previous anti-HER2 treatment. In addition, there is evidence of HER2 discordance between primary and metastatic tumours and between tumours before and after first line chemotherapy for metastatic disease (with or without trastuzumab)[17-21]. Meanwhile, high incidence of heterogeneous HER2 overexpression in gastric cancer might affect activity of T-DM1 because the non-cleavable linker does not allow for bystander activity[22,23]. Future clinical trials are warranted to clarify the role of T-DM1 either combining with chemotherapy or with immunotherapy in advanced gastric cancer. The phase I/II TRAX-HER2 trial (NCT01702558) of capecitabine +/- T-DM1 has recently completed,which demonstrated that the combination of T-DM1 and capecitabine is tolerable in HER2-positive advanced breast and gastric cancer patients[24]. However, there was no statistically significant difference in ORR between patients with metastatic breast cancer who received T-DM1 + capecitabine vs. T-DM1 alone(44.4% vs. 36.3%; P = 0.336)[25]. Two phase II clinical trials (NCT03418558 and NCT03225937) are currently recruiting patients with metastatic colorectal cancer to evaluate the anti-tumour benefits of T-DM1 alone or in combination with programmed cell death receptor 1 (PD-1) inhibitor pembrolizumab.

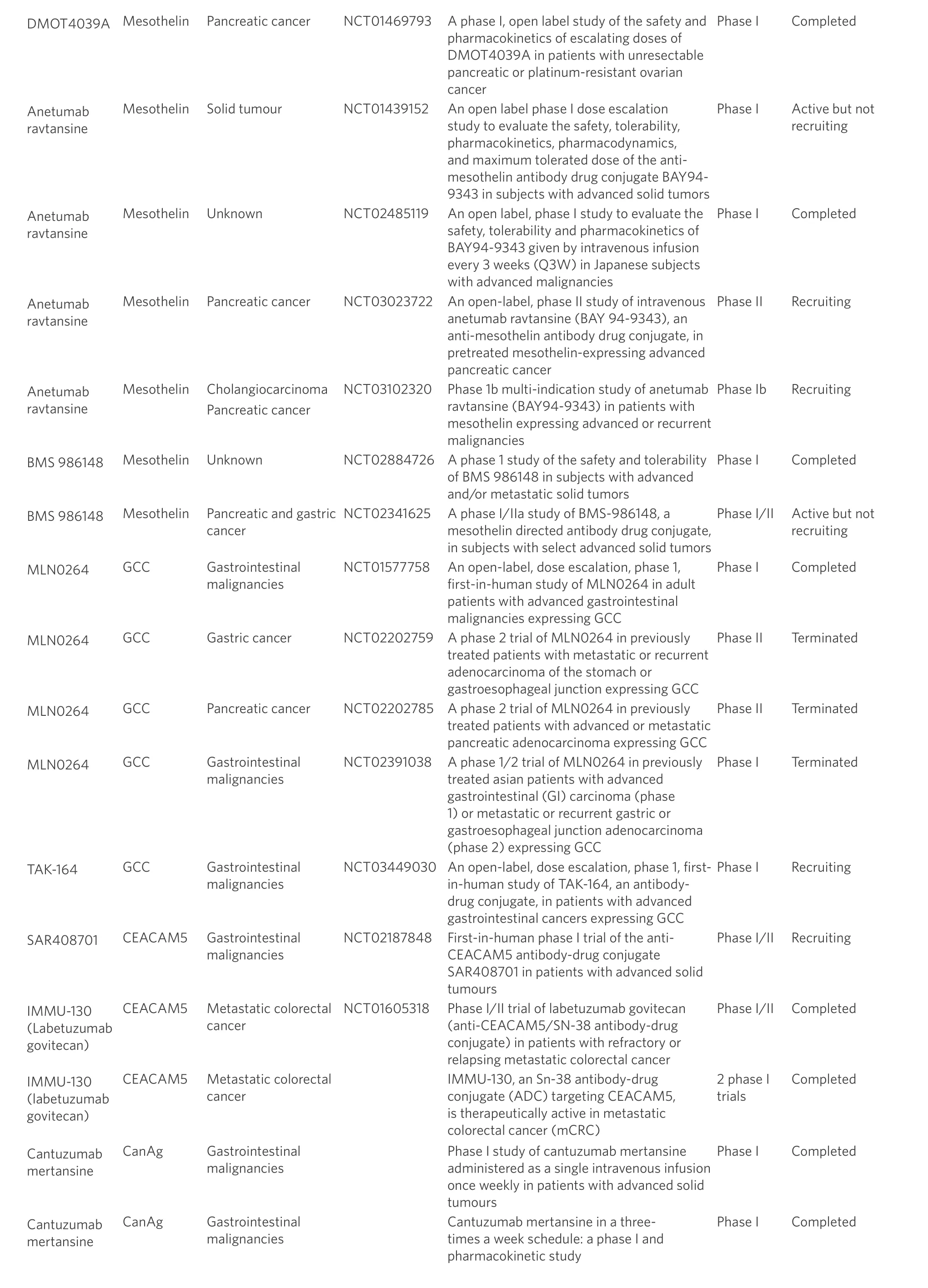
Table 1. Antibody drug conjugate trials in gastrointestinal cancers
?

HER2: growth factor receptor 2; GCC: guanylyl cyclase C
DS-8201A
DS-8201a is a new HER2-targeted ADC, which conjugates a humanized anti-HER2 antibody to a topoisomerase I inhibitor, deruxtecan, by a cleavable peptide-linker[26]. Unlike T-DM1, DS-8201a has a potent bystander killing effect due to its cleavable linker, which is beneficial in treating heterogeneous HER2 overexpressing tumours, such as gastric cancer. Also DS-8201a has a higher drug to antibody ratio (7 or 8) compared to that of T-DM1 (3 or 4). DS-8201a demonstrated promising antitumor activity in T-DM1 resistant HER2 expressing gastric cancer in pre-clinical trials because of these unique characteristics[27,28].A phase I multi-centre, dose escalation study (NCT02564900) reported in 2016 that DS-8201a was well tolerated with satisfactory safety profiles. Only 14% of patients (n = 3/22) experienced more than grade 3 AE.The most common AE were GI and haematological toxicities. A dose of 6.4 mg/kg every three weeks was selected as the recommend phase II dose (RPIID). However there were no dose limiting toxicities identified in this study and the MTD was not established. An ORR of 35% and a disease control rate (DCR) of 90%were reported across several tumour types including breast and gastric cancer[29,30]. An updated report of this study (NCT02564900) in February 2018 further confirmed that ORR and DCR were 44% (16/36) and 78%(28/36) respectively in HER2 expressing gastric cancer patients. It also reported that 83% of subjects were experiencing tumour shrinkage[31]. In addition, DS-8201a showed efficacy in patients with HER2 expressing non-breast and non-gastric malignancies. ORR (including outcomes awaiting confirmation) and DCR were 33% and 91% respectively in 12 evaluable patients. Common side effects from DS-8201a were nausea,reduced appetite, vomiting and reduced platelet count[32]. The study of DS-8201a progressed to a phase II trial (NCT03329690) to evaluate superiority of DS-8201a vs. either irinotecan or paclitaxel monotherapy in gastric cancer. Another phase II trial (NCT03384940) has also commenced to investigate benefit of DS-8201a in advanced colorectal cancer.
NEW GENERATION HER2 TARGETED ADCS
XMT-1522 employs a novel human anti-HER2 antibody conjugated with an auristatin (microtubule polymerization inhibitor)-derivative payload, Auristatin F-Hydroxypropylamide, via a biodegradable hydrophilic polymer, which increased drug to antibody ratio of XMT-1522 to 10-15[33]. In pre-clinical trials,XMT-1522 exhibited anti-tumour efficacy in highly HER2-expressing gastric cancer models. Tumour regression was seen either with XMT-1522 alone or with the XMT-152/trastuzumab/pertuzumab triple combination[34]. A dose escalation phase I study (NCT02952729) in patients with HER2-expressing breast,lung and gastric cancers demonstrated that XMT-1522 was well-tolerated up to the 21.3 mg/m2dose level with only grade 1 or 2 AE observed. The common AE were impaired liver function, fatigue, nausea,vomiting, reduced appetite and headache. MTD was not established in this study as no dose limiting toxicity was identified. As of February 1st 2018, there are 19 patients enrolled where DCR was 83% (5/6) for patients dosed at 16 or 21.3 mg/m2. Two stable disease (SD) outcomes were seen in gastric cancer patients and others in breast cancer patients. Dose escalation continues with more results expected[35].
MEDI-4276 is another ADC approach to HER2 expressing tumours. MEDI-4276 is a biparatopic ADC that targets two different epitopes on HER2, with site-specific conjugation to a tubulysin-based microtubule inhibitor. MEDI-4276 utilizes a cleavable linker that enables it to mediate bystander killing activity[36]. Preclinical trials indicated that MEDI-4276 has anti-tumour activity in vitro, including in T-DM1 resistant cells.A phase I/II dose escalation trial (NCT02576548) had recruited 43 patients with HER2-expressing breast or gastric cancers by November 2017. MTD at 0.9 mg/kg showed dose limiting toxicity of grade 3 elevated alanine aminotranferase (ALT)/aspartate aminotransferase (AST) or grade 3 diarrhea. Common AE were nausea, fatigue, vomiting and elevated ALT/AST. In this study, MEDI-4276 exhibited anti-tumour activity with 1 complete response, 1 partial response (PR) and 12 (28%) SD[37]. A further report is awaited.
SYD985 consists of trastuzumab conjugated to the DNA alkylating agent duocarmycin via a cleavable linker. Multiple pre-clinical trials strongly suggested that SYD985 had extended anti-tumour activity in HER2 low-expressing or HER2 negative breast, gastric, ovarian or uterine cancer cell lines, when compared directly to T-DM1. Unlike T-DM1, SYD985 is able to induce efficient bystander killing effect in vivo, which contributes to significantly improved anti-tumour activity of SYD985 when compared to T-DM1[38-41]. A phase I dose expansion cohort study (NCT02277717) showed promising efficacy of SYD985 in heavily pretreated breast cancer patients, including hormone-receptor positive and triple negative breast cancer.The MTD was 2.4 mg/kg with a manageable safety profile, mainly characterized by grade 1/2 AE of fatigue,dry eyes, conjunctivitis and increased lacrimation. Dose limiting toxicity included grade 3/4 AE including neutropaenia and conjunctivitis. The ORRs were 27% and 40% respectively in hormone-receptor positive and triple negative breast cancer patients; several patients continue on SYD985. Further dose expansion date is pending in gastric cancer patients[42].
ARX-788 is a site-specific ADC conjugating amberstatin (tubulin inhibitor) to a HER2 antibody at two specific sites via a non-cleavable linker. Pre-clinical studies reported that ARX-788 demonstrated increased anti-tumour activity in breast, ovarian, lung and gastric cancer xenograft models compared to T-DM1.Unlike T-DM1, the site-specific conjugated form of ARX-788 has increased stability of up to 3 weeks which improved its therapeutic window and anti-tumour activity[43]. There are currently two dose escalation phase I trials recruiting patients with HER2-expressing breast and gastric cancers (NCT03255070 and NCT02512237)with the intention to identify a safety profile of ARX-788 and potential anti-tumour efficacy.
ADCT-502 is made of an engineered version of trastuzumab, site-specifically conjugated to a highly cytotoxic pyrrolobenzodiazepine-based drug, tesirine, via a protease cleavable linker. It has a drug to antibody ratio of 1.7 and demonstrates bystander killing effect in vitro. There is superiority in anti-tumour effect of ADCT-502 compared to T-DM1 in various tumour xenografts, including those with low HER2 expression levels[44].ADCT-502 is currently in a phase I dose escalation trial (NCT03125200) involving patients with HER2 expressing advanced solid tumours, including gastroesophageal cancer.
These new generation HER2 targeted ADCs showed superior anti-tumour activity compared to T-DM1 in various pre-clinical trials, even in HER2 low-expressing or HER2 negative cancer models. Consequently these new generation of HER2 targeted ADCs provide a potential treatment option not only in patients who become resistant/refractory to T-DM1, but also in patients whose tumours express low-level or no HER2,and are not eligible for treatment with T-DM1.
MESOTHELIN TARGETED ADCS
Mesothelin is a tumour differentiation antigen that is highly expressed in certain human malignancies which includes 86%-100% of pancreatic ductal adenocarcinomas[45], 97% of ovarian adenocarcinomas and 89%-100% of mesotheliomas. The limited expression of mesothelin in essential human tissue and high expression in certain cancers make mesothelin a good target for cancer therapy, such as in pancreatic cancer[46]. Several anti-mesothelin antibody based treatments are under clinical investigation, which include vaccines, T-cell therapy and ADC therapy[47]. Amatuximab is the first monoclonal mesothelin targeted antibody. In a phase II clinical trial (NCT00570713), Amatuximab combined with gemcitabine showed no clinical benefit in patients with pancreatic cancer when compared to placebo groups[48]. Further investigation of mesothelin targeted therapy, such as new generation anti-mesothelin antibodies and ADCs, are in progress.
DMOT4039A is the first generation of mesothelin targeted ADCs. DMOT4039A consists of humanized anti-mesothelin mAb conjugated to an antimitotic agent, monomethyl auristatin (MMAE) via a protease cleavable linker. DMOT4039A has a drug to antibody ratio of approximately 3.5:1. A pre-clinical trial showed tumour growth inhibition in pancreatic cancer xenografts treated with DMOT4039A[49]. A phase I study of DMOT4039A (NCT01469793) in patients with unresectable pancreatic cancer or platinum resistant ovarian cancer demonstrated that DMOT4039A has antitumor activity with an acceptable safety profile. In this study,the RPIID level was determined to be either 2.4 mg/kg three weekly or 1.0 mg/kg weekly. Dose limiting toxicities included grade 3 hyperglycaemia and hypophosphatemia. eight percent (2/26) of patients with pancreatic cancer on the three weekly regime achieved PR and 35% (9/26) of patients had SD as best response with a median duration of 5.7 months. The low response rate might be explained by inherent variability in assessing small cohorts or heterogeneous expression of mesothelin in pancreatic cancer[50]. Therefore,DMOT4039A warrants further clinical trials in combination with either chemotherapy or immunotherapy in patients with pancreatic cancer.
Anetumab ravtansine, also called BAY 94-9394 is another potent mesothelin targeted ADC. Anetumab ravtansine is composed of human anti-mesothelin antibodies conjugated to the microtubule polymerase inhibitor, DM4, through a disulphide bond. Pre-clinical trials showed that anetumab ravtansine exhibited antiproliferative activity with bystander effect in mesothelin positive cancer cell lines, which included pancreatic cancer and patient-derived pancreatic cancer xenograft models[51]. In a phase I trial (NCT01439152),MTD of anetumab ravtansine was established at 6.5 mg/kg given every three weeks. The AE included reversible keratitis, asymptomatic liver impairment and GI disorders[52]. A parallel trial (NCT02485119)enrolling Japanese patients showed similar results. Several phase I/II clinical trials are actively recruiting patients presenting with different malignancies. A phase Ib study (NCT03102320) is recruiting patients with mesothelin expressing advanced cholangiocarcinoma or advanced pancreatic adenocarcinoma. This study aims to test the MTD of anetumab ravtansine in combination with either cisplatin or gemcitabine for cholangiocarcinoma or adenocarcinoma of the pancreas[53]. A phase II, multicentre, non-randomized trial(NCT03023722) is evaluating the efficacy of anetumab ravtansine in patients with pre-treated mesothelinexpressing advanced pancreatic cancer[54].BMS-986148 is another ADC that targets to mesothelin. Limited information is known regarding this ADC.BMS-986148 may be related to MDX-1204, which is a conjugation of an anti-mesothelin antibody to the alkylating agent, duocarmycin[55]. A phase I study (NCT02884726) in a single centre in Japan was completed and will provide reports about safety and tolerability of BMS-986148 in patients with advanced and/or metastatic solid tumours. A phase I/II clinical trial (NCT02341625) is currently active, but not recruiting.The goal is to determine the MTD and safety profile of BMS-986148 administered alone or in combination with nivolumab in patients with mesothelioma, non-small cell lung cancer, ovarian cancer, pancreatic cancer and gastric cancer.
GCC TARGETED ADCS
GCC is a heat-stable enterotoxin receptor, which is only expressed on intestinal epithelial cells but not extra-GI cells. Previous studies showed that GCC is highly expressed in 60% to 70% of pancreatic, gastric and oesophageal cancers and 95% of primary/metastatic colorectal cancers[56,57]. As a consequence, GCC provides another feasible target for oncology treatment in GI malignancies.
TAK-264 (also called MLN0264) is an ADC that consists of a fully human anti-GCC mAb conjugated, via a protease-cleavable linker, to the microtubule-disrupting agent MMAE. Pre-clinical trials demonstrated that TAK-264 has selective antitumor activity in GCC-expressing colorectal and pancreatic cancer lines and xenografts[57,58]. A multicentre, dose escalation phase I study (NCT01577758) suggested that the MTD of TAK-264 is 1.8 mg/kg every 3 weeks. At this dose level, TAK-264 has a manageable safety profile with common AE including nausea, reduced appetite, diarrhea and fatigue. In this study, forty-one patients were enrolled,including 35 (85%) with metastatic colorectal cancer. Overall, thirty-nine patients were response-evaluable;three patients experienced SD; 1 patient with gastric adenocarcinoma had a PR[59]. A parallel phase I study(NCT02391038) in Asian patients demonstrated a similar safety profile with TAK-264. However, in this study, none of the patients experienced a dose limiting toxicity and the MTD was not reached up to 1.8 mg/kg three weekly. All 12 patients were response evaluable; three patients had SD[60]. Preliminary evidence that showed clinical benefit of TAK-264 in phase I trials warranted pursuit of a phase II (NCT02202785) clinical study. This study tested efficacy of TAK-264 in patients with previously treated advanced or metastatic pancreatic cancer. Forty-three patients were enrolled and treated with 1.8 mg/kg TAK-264 every three weeks.Ninety-one percent patients showed measurable response; the ORR was 3% including 1 patient having PR.Nine patients (23%) achieved SD based on investigator assessment. The study was terminated early due to limited benefit[61]. A phase I study that recruited patients with gastric/gastroesophageal junction cancers(NCT02202759) showed similar results. Among 38 patients enrolled, thirty-six patients were response evaluable; the ORR was 6% including 2 patients (6%) having achieved PR. Fifteen patients (42%) achieved SD. The study was terminated after stage I due to low patient response[62]. However, no clear mechanisms were identified to explain the low efficacy of TAK-264. One reason might be the large size of TAK-264, which reduced internalization and consequently results in low activity. GCC remains a favorable target for ADC development for patients with high GCC expression.
A novel GCC targeted ADC, TAK-164, has progressed to the early phases of clinical trials. TAK-164 is comprised of an anti-GCC mAb conjugated to the DNA alkylating agent, DGN549 (also known as IGN-P1).Pre-clinical trials demonstrated that TAK-164 had potential antitumor activity in colorectal or other GI xenografts[63]. Currently, a phase I trial (NCT03449030) is recruiting patients with GCC expressing advanced GI cancers.
CEACAM5 TARGETED ADCS
CEACAMs are a large family of twelve glycosyl phosphatidyl inositol anchored glycoproteins involved in cell adhesion and cell signaling during complex biological processes such as cancer progression, in flammatio angiogenesis, and metastasis. CEACAM5 has been recognized as a tumour marker of colorectal cancers for the last 50 years and is highly expressed at the surface of tumour cells, including more than 80% of colorectal cancer cells. CEACAM5 has been recently exploited as a target for immunotherapy and vaccine-based therapeutics. In the animal models of colorectal and pancreatic cancers, antibodies against CEACAM5 have shown their potential effectiveness in cancer treatment[64]. Therefore, CEACAM5 is another potential target in ADCs development.
SAR408701 (SAR) is an ADC comprising an anti-CEACAM5 antibody, a cleavable N-succinimidyl 4-(2-pyridyldithio) pentanoate linker and the microtubule-targeting mitosis inhibitor, DM4. Pre-clinical studies have shown promising anti-tumour efficacy with SAR in colorectal cancer, lung and gastric xenografts[65]. A phase I study (NCT02187848) investigated the safety and pharmacokinetics of SAR in patients with GI tumours expressing CEACAM5. In this study, SAR showed an acceptable safety profile with the MTD established as 100 mg/m2. The most commons AE were fatigue, visual/eye changes and GI disturbances. Twelve colorectal cancer patients were tested as CEACAM5 positive, three of which sustained PR (lasting > 4 months)[66]. A dose expansion trial in CEACAM5-expressing solid tumours (including colorectal and gastric cancers) is ongoing with further results on the way (NCT02187848).
Labetuzumab govitecan (IMMU-130) is a CEACAM5 targeted ADC which conjugates the CEACAM5 antibody labetuzumab to the active metabolite of irinotecan, SN-38, through a proprietary linker.Labetuzumab govitecan showed improved efficacy and reduced toxicity compared to irinotecan in preclinical colorectal cancer xenografts[67-69]. Two phase I studies investigating the activity of labetuzumab govitecan in patients with metastatic colorectal cancer were reported in a conference abstract in 2014. In both trials, eligible patients had been pre-treated with standard chemotherapy (such as irinotecan) and had an elevated CEA (> 5 ng/dL). In the first study, labetuzumab govitecan was given every two weeks; the MTD was 16 mg/kg. One patient (out of 8) received 18 cycles of treatment and achieved PR with a 40.6% reduction in liver and lung lesions, lasting for 4.7 months. In the second study, MTD was the same when labetuzumab govitecan was given 4 mg/kg twice weekly. Three/six patients experienced PR with 40% tumour shrinkage.In these two phase I trials, labetuzumab govitecan was well tolerated with the most common AE including neutropaenia and diarrhea[70].
A phase I/II, multicentre dose-finding trial (NCT01605318) investigated the therapeutic index of labetuzumab govitecan in patients with relapsed or refractory metastatic colorectal cancer. Eighty-six patients were recruited with an elevated serum CEA (> 5 ng/mL) and had progressed after a median of five prior treatments (including irinotecan). Weekly doses (8 or 10 mg/kg) or twice weekly doses (4 or 6 mg/kg) with 1 week breaks were demonstrated to be tolerable. The most common AE were neutropaenia,leukopenia, anaemia and diarrhea. In this trial, thirty-eight percent patients (27/72) achieved a reduction in tumour size with one patient achieving a sustained PR lasting for 2.7 years. Fifty-eight percent (42/72)patients achieved SD with 33% (24/72) of these outcomes lasting more than 4 months. Median PFS for all 86 patients was 3.6 months (95% CI 2-4 months) and median OS was 6.9 months (95% CI 5.7-7.8). Compared to the PFS and median OS of regorafenib (1.9 and 6.4 months, respectively), which is currently recommended as third line treatment for relapsed or refractory metastatic colorectal cancer, labetuzumab govitecan provides another option in the third-line setting[71]. Labetuzumab govitecan also showed less toxicity than irinotecan,particularly with regard to diarrhea. Combination approaches of labetuzumab govitecan with other agents should be investigated; a pre-clinical study has demonstrated an enhanced anti-tumour effect in colorectal cancer cells of labetuzumab govitecan combined with bevacizumab[72].
OTHER ADCS IN GI CANCERS
Cantuzumab mertansine is an ADC combining huC242, a highly selective mAb targeting CanAg, with DM1 via a disulphide linker. CanAg is a transmembrane glycoprotein that is highly expressed in pancreatic cancer, cholangiocarcinoma and colorectal cancer[73]. Cantuzumab mertansine was highly effective against CanAg positive tumours in pre-clinical studies that showed complete responses in CanAg positive gastric, colon, pancreatic and non-small cell lung cancer xenografts[74]. Three phase I studies have assessed cantuzumab mertansine in solid tumours, including GI cancer patients. AE included fatigue, deranged liver function tests and GI disturbances. Best response was SD, lasting 4-5 months on average[73,75,76]. Cantuzumab mertansine did not progress to phase II study for unknown reasons.
PF-06263507 is an ADC comprising humanized anti-5T4 antibodies (huA1), a non-cleavable linker and a monomethylauristatin F payload (a microtubule polymerization inhibitor)[77]. 5T4 is a surface antigen expressed in many tumour types including pancreatic, oesophageal, colorectal and gastric cancers. High levels of 5T4 expression have been associated with poorer clinical outcomes in colorectal, ovarian and gastric cancers[78-81]. In a phase 1 study, twenty-six patients with solid tumours (including GI cancers) were treated with PF-06263507 with the MTD determined to be 4.34 mg/kg. 5T4 expression was not assessed. No ORRs were observed; two patients achieved SD. The most common AE were fatigue, photophobia, dry eyes and GI disturbances[77]. Pre-clinical studies have since demonstrated how 5T4 positive cells can down-regulate expression of 5T4 in response to 5T4 targeting ADCs, leading to resistance over time[82-84]. Pre-clinical studies have also revealed enhanced anti-tumour activity of combination treatments involving PF-06263507[85,86].
CONCLUSION AND FUTURE PROSPECTS
GI malignancies represent the most common cancers and are a leading cause of cancer-related death worldwide. ADCs are a rapidly growing cancer treatment that are expected to selectively deliver cytotoxic drugs to cancer cells for increased efficacy, while minimizing toxicity to normal healthy cells. In this review,we have outlined pre-clinical and clinical evidence of current popular ADCs trials in GI malignancies,including HER2, mesothelin, GCC and CEACAM5 targeted ADCs. However, none of these ADCs have advanced into clinical practice for treating GI cancers.
Future strategies to investigate the efficacy of ADCs should assess combinations with other oncology modalities including chemotherapy, radiotherapy, immune checkpoint inhibitors or targeted therapy.Most clinical trials of ADCs in GI malignancies to date have attempted to demonstrate superiority as monotherapy. However, their higher tolerability and safer toxicity profiles, when compared to cytotoxic chemotherapy, indicate that they may be suitable to combine with other agents. Although the TRAXHER2 study did not show superiority of the T-DM1/capecitabine combination compared to T-DM1 alone in patients with metastatic breast or gastric cancer, many other chemotherapy/ADC combinations are under consideration to enhance anti-tumour effects of ADCs. Furthermore, ADCs with DNA damaging agents(especially microtubule inhibitors) as the payload have the potential to enhance and sustain anti-tumour effects when combined with immune checkpoint inhibitors such as anti-PD1 antibodies (Pembrolizumab,Nivolumab) or anti-programmed death ligand 1 antibodies (atezolizumab). There is accumulating preclinical evidence to suggest that DNA damaging agents can promote anti-tumour immune responses by increasing antigen presentation, activating dendritic cells and promoting immunogenic cells death[87]. Preclinical trials proved that T-DM1 renders HER2 positive breast cancer cells highly susceptible to PD-1 blockade[88]. An active phase I/II clinical trial (NCT02318901) is evaluating the potential of T-DM1 in combination with pembrolizumab in advanced breast, gastric, oesophageal and colorectal cancers. Finally,HER2 over expression was identified as a factor contributing to radio-resistance in breast cancers. Radiation may result in a more potent anti-tumour effect when combined with anti-HER2 therapy. Further exploration of concurrent administration of T-DM1 with radiotherapy is warranted in GI cancers[89]. The challenges of combining ADCs with other modalities includes choice of ADC with other cytotoxic agents, dose of compounds, toxicity of combination and scheduling/sequencing of combinations.
Another major trend in ADC research is to design new generations of ADCs with increased homogeneity,stability and potency through the large variety of ADC technologies developed over the past decade. The current third generation of ADCs has significantly benefited from the engineering of IgG molecules,which are well suited for drug conjugation and enhance the homogeneity and therapeutic index of ADCs.Moreover, the first biparatopic ADC, MEDI-4276, targets two non-overlapping epitopes on the same HER2 antigen and can thus potentially improve drug delivery to tumours while reducing drug exposure to normal tissue. A variety of new formats of mAbs are under investigation in pre-clinical stages, providing new opportunities to develop antibody-dual-drug conjugates or nanobodies/single chain fragment/peptide-drug conjugates in the future. In addition, diversification of linking strategies such as cleavable/non-cleavable linkers and charged/non-polar linkers have been utilized in linker design to balance the need for stability in the circulation and efficient cleavage upon delivery into tumour cells, therein enhancing or reducing bystander effects. Several linking strategies are also under investigation to increase solubility and drugantibody ratio of ADCs[3].
Finally, patient selection strategies in the clinical study of ADCs can make the difference between success and disappointment. The level of antigen expression on cancer cells' surface is a key parameter in predicting the likelihood of the amount of payload delivered to a cancer cell in a tumour. Thus, selecting patients whose target antigen density (on their cancer cells' surface) is above a threshold level is important to optimising ADC activity. However, one of the challenges to meet this criterion is identifying the presence of target antigens in real time and by minimally invasive techniques. Several strategies to develop new diagnostic tests to assess the level of cell target antigens are under investigation. For example, 89Zr radiolabeled trastuzumab positron emission tomography scans are able to demonstrate, in real time, HER2 expression levels with the advantage of being relatively non-invasive[90]. Circulating DNA can also predict HER2 status, reducing the need for invasive biopsies. There is clearly much yet to explore in the future for biomarkers to predict whether patients will respond to ADC treatment.
In summary, incorporating ADCs into future GI malignancy treatment offers exciting opportunity for better outcomes for cancer patients. Future strategies will focus on optimal application of ADCs, especially in establishing the best combination modalities for treating different GI tumours, designing new generation of ADCs with high potency and stability as well as co-developing biomarker detection technologies for better patient selection.
DECLARATIONS
Acknowledgement
All authors would like to acknowledge National Health Service funding to the National Institute for Health Research Biomedical Research Centre at the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research.
Authors' contributions
Wrote the manuscript: Wu X, Kilpatrick T
Edited, reviewed and approved the manuscript: Chau I
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Chau I is on the advisory board for Eli-Lilly, Bristol Myers Squibb, MSD, Bayer, Roche, Merck-Serono, Five Prime Therapeutics and Astra-Zeneca. He has received research support from Eli-Lilly, Janssen-Cilag, Sano fi
Oncology, Merck-Serono and honorarium from Eli-Lilly.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
杂志排行
Cancer Drug Resistance的其它文章
- Meeting Abstracts of the BACR conference:response and resistance in cancer therapy
- What really matters - response and resistance in cancer therapy
- Wilms' tumor gene (WT1) is strongly expressed in high-risk subsets of pediatric acute lymphoblastic leukemia
- Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer
- Epigenetics, a key player of immunotherapy resistance
- Acknowledgement to reviewers of Cancer Drug Resistance in 2018