大麦黄条点花叶病毒的分布及其分离物的遗传多样性
2018-02-04杨菲张爱红孟凡思霍良占李希望邸垫平苗洪芹
杨菲,张爱红,孟凡思,霍良占,李希望,邸垫平,苗洪芹
大麦黄条点花叶病毒的分布及其分离物的遗传多样性
杨菲,张爱红,孟凡思,霍良占,李希望,邸垫平,苗洪芹
(河北省农林科学院植物保护研究所/河北省农业有害生物综合防治工程技术研究中心/农业部华北北部作物有害生物综合治理重点实验室, 河北保定 071000)
【目的】明确大麦黄条点花叶病毒(,BYSMV)在中国北方小麦主产区的分布及其种群遗传多样性,为病害流行预警和防控提供理论依据。【方法】2008—2016年,在河北、山东、江苏、安徽、河南、陕西和山西等7个省66个县/市/区田间,采集了864份疑似病毒病症状的植物样品。提取样品总RNA,利用一步法三重RT-PCR技术检测样品中的BYSMV、水稻黑条矮缩病毒(,RBSDV)和北方禾谷花叶病毒(,NCMV)。利用RT-PCR扩增获得BYSMV的L和N基因片段,克隆并测定核苷酸序列,应用MEGA、DnaSP和PAML等软件分析BYSMV分离物的系统进化和遗传多样性特征。【结果】从48个县/市/区采集的336份样品中检测到BYSMV,检出率为38.89%,该病毒主要分布于陕西、河北、山西和山东,另外,河南及安徽北部亦有分布,江苏徐州和邳州仅局部发生。基于BYSMV的L、N基因序列构建的系统发育树均可将分离物划分为2个亚组,亚组I中的分离物其来源涉及全部7个省份,而亚组II中的分离物仅来自陕西和山西2个省,基于L基因序列系统发育分析表明亚组II分离物与伊朗的分离物亲缘关系较近,BYSMV的遗传分化与分离物的地理来源相关,而与寄主植物、发生时间无明显相关性。运用RDP程序包的7个软件进行基因重组分析显示没有支持重组的证据。选择压力分析显示,亚组内和亚组间的ω(dN/dS)值(0.02—0.19)远小于1,表明群体正承受净化选择。L和N基因的单倍型多样性(Hd)值(0.90909和0.99524)均大于0.5、核苷酸多样性(π)值(0.01324和0.01224)均高于0.005,表明中国BYSMV群体遗传多样性丰富。基于L和N基因片段的遗传分化研究显示,东部和西部群体的遗传分化系数(F)值(0.32201和0.37326)均大于0.25,且统计检验差异显著,表明东部和西部的BYSMV群体严重分化;基因流(Nm)值(0.53和0.42)均小于1,说明有限的基因流是促使群体发生遗传分化的主要原因。【结论】BYSMV在中国北方小麦主产区分布广泛,河北、山东、江苏、安徽、河南、陕西和山西等地均有不同程度的发生。BYSMV群体具有丰富的遗传多样性,且东部和西部群体之间存在严重的遗传分化。
大麦黄条点花叶病毒; 系统发生; 遗传多样性; 选择压力; 遗传分化
0 引言
【研究意义】大麦黄条点花叶病毒(,BYSMV)是灰飞虱()传播的一种弹状病毒,侵染小麦、大麦、玉米、高粱等近30种禾本科植物[1],在小麦上引起的病害十分严重,可造成植株严重矮化,叶片对生、细窄、扭曲和黄化等症状[2]。小麦是中国重要粮食作物,2014年笔者实验室首次报道了在河北保定、石家庄麦田发现大麦黄条点花叶病毒[3],该病毒的传毒介体灰飞虱在小麦种植区广泛分布,因此,调查明确大麦黄条点花叶病毒在北方小麦主产区的发生情况,为病害防控预警提供理论依据,对于保障粮食安全生产具有重要意义。【前人研究进展】大麦黄条点花叶病毒最初在灰飞虱体内分离发现[4],而后澳大利亚[5]、摩洛哥[6]、伊朗[7]、土耳其[8]、叙利亚[9]及阿根廷[10]等地相继报道该病毒,其在伊朗引起严重的小麦和谷子病害[7]。大麦黄条点花叶病毒是细胞质弹状病毒属成员,其基因组为负单链RNA,Yan等[11]利用小RNA深度测序和组装技术获得了全长约12.7 kb的基因组序列,基因结构为3′ leader-N-P-3-4/5-6-M-G-9-L-5′ trailer,与其他弹状病毒类似,分别编码核衣壳蛋白(nucleocapsid protein,N)、磷蛋白(phosphoprotein protein,P)、基质蛋白(matrix protein,M)、糖蛋白(glycoprotein protein,G)和聚合酶蛋白(polymerase protein,L),与北方禾谷花叶病毒(,NCMV)相应氨基酸序列一致性为35%—62%。除5个基本的结构蛋白以外,还编码5个功能各异或功能未知的辅助蛋白(accessory protein),在基因P和M之间有3个转录单位,其中gene 5是一个嵌套基因,编码疏水蛋白,通过核糖体漏扫机制进行蛋白翻译,这种非常规的基因表达策略在其他植物弹状病毒中未见报道,而在动物弹状病毒中较为常见。Almasi等[12]还报道了大麦黄条点花叶病毒伊朗分离物的L、P和G基因全长序列(https:// www.ncbi.nlm.nih.gov/)。植物病毒,特别是RNA病毒具有快速变异的能力。由于RNA聚合酶缺乏校对机制,RNA病毒具有很高的突变率,据估计碱基替换率为10-4[13-14]。基因变异导致病毒宿主范围和病害症状发生变化,甚至进化为“新病毒”引起更严重的病害。研究表明,水稻条纹病毒(,RSV)[15]、大麦黄矮病毒(,BYDV)[16]和水稻黑条矮缩病毒(,RBSDV)[17]种群结构表现遗传多样性,重组是基因变异的主要方式。莴苣坏死黄化病毒(Lettuce necrotic yellows virus,LNYV)、草莓皱缩病毒(Strawberry crinkle virus,SCV)和茄子斑驳矮缩病毒(Eggplant mottled dwarf virus,EMDV)3种植物弹状病毒田间分离物的L及N基因核苷酸和推导的氨基酸序列存在相当多的遗传变异性,通过对莴苣坏死黄化病毒N基因的分析,澳大利亚和新西兰的分离物被划分为2个亚组[18];根据对L基因片段的变异分析草莓皱缩病毒被分为2个亚组[19];Pappi等[20]研究了不同地理起源的茄子斑驳矮缩病毒草本和木本植物分离物的N、X、P、Y、M、G基因编码序列,不同分离物基于其地理来源可分成3个亚组。【本研究切入点】中国小麦种植区域极为广泛,生态环境多样,目前大麦黄条点花叶病毒在不同地区的发生动态尚不明确,小麦病害流行预警和防控策略缺乏理论依据。【拟解决的关键问题】采用分子生物学方法鉴定麦田灰飞虱传播的病毒种类,并对大麦黄条点花叶病毒的基因序列进行分析,明确大麦黄条点花叶病毒在小麦种植区的分布及遗传多样性特征,为有效防控该病毒引起的病害打下基础。
1 材料与方法
1.1 试验材料
2008—2016年分别于河北、山东、江苏、安徽、河南、陕西和山西7省田间调查,采集植株矮化、旗叶发黄等疑似感染病毒的小麦、玉米、杂草叶片,保存于-70℃冰箱。部分病株样品采集时间、采样地点、寄主植物等信息列于表1。病毒检测阳性对照是通过室内人工接种大麦黄条点花叶病毒、水稻黑条矮缩病毒和北方禾谷花叶病毒而获得表现症状的小麦病株,阴性对照为未接种病毒的健康小麦。
试剂及仪器:RNA提取试剂、一步法RT-PCR试剂盒购自康为世纪公司,琼脂糖凝胶回收试剂盒购自TIANGEN公司,cDNA第一链合成试剂盒、Dream Taq DNA聚合酶、dNTP等PCR试剂均购自Thermo Scientific公司。其余试剂为国产分析纯。Veriti PCR仪,美国Thermo Fisher Scientific公司;DYY-12型电泳仪,北京六一生物科技有限公司;GBOXF3凝胶成像分析系统,英国Syngene公司。
1.2 总RNA提取方法
参照康为世纪TRIzon试剂盒说明书提取样品总RNA,经NanoDrop ND2000分光光度计测定浓度,于-20℃保存备用。
1.3 病毒检测
采用大麦黄条点花叶病毒、水稻黑条矮缩病毒和北方禾谷花叶病毒特异性引物BYSMVF:5′-TCCGCA GGTAGACGCCAAGAAG-3′和BYSMVR:5′-CGCAG TCCCAGTCAGAAAGGTG-3′、RBSDVF:5′-TTGGGA TTTGGTCGTCGTAGAGCC-3′和RBSDVR:5′-AACT TCGCATGAACTGGGGTTGTC-3′、NCMVF:5′-AAA CAGAGCTTCACGGAGACTTGG-3′和NCMVR:5′-A ACACTAACCCTCCCACGTCTGTA-3′进行RT-PCR反应检测样品中的3种病毒。总体积12.5 μl的反应体系中包括2×OneStep RT-PCR Buffer 6.25 μl,终浓度为0.1 μmol·L-1的引物各0.125 μl,HiFi-MMLV OneStep RT-PCR EnzymeMix 0.25 μl,模板样品总RNA 1 μl,无核酸酶ddH2O 4.25 μl。反应程序:45℃反转录30 min,95℃预变性5 min,94℃变性30 s,54℃退火30 s,65℃延伸1 min,进行30个循环,65℃终延伸10 min。阳性对照为混合的3个阳性对照的总RNA,阴性对照为未接种的健康小麦总RNA。
1.4 大麦黄条点花叶病毒L、N基因引物的设计与合成
根据已报道的大麦黄条点花叶病毒河北分离物(GenBank登录号:KM213865)的L、N基因序列分别设计了1对引物。扩增L基因片段的上下游引物序列分别位于L基因的2个保守结构域上,产物全长637 bp,中间387 bp的序列为非保守区;扩增N基因片段的上下游引物分别位于N基因的两端,可扩增获得N基因的全长序列。引物由上海生工生物科技有限公司合成,序列信息见表2。
1.5 大麦黄条点花叶病毒L、N基因RT-PCR扩增及测序
第一链cDNA合成参照Thermo Scientific合成试剂盒说明书进行。以cDNA为模板进行PCR扩增,反应体系为20 μL:反转录产物1 μL,上下游特异性引物(10 mmol·L-1)各0.5 μL,10×Dream Taq Buffer 2 μL,dNTP(10 mmol·L-1)0.2 μL,Dream Taq DNA聚合酶0.2 μL及ddH2O 15.6 μL。反应程序:95℃预变性5 min,95℃变性30 s,50℃退火30 s,72℃延伸110 s,进行30个循环,72℃终延伸10 min。反应结束后取5 μL扩增产物,经1.0%琼脂糖凝胶电泳检测,利用TIANGEN公司的琼脂糖凝胶回收试剂盒对目的片段进行切胶纯化。参照TaKaRa公司pMD18-T Vector说明书,将纯化产物连接至pMD18-T载体,转化大肠杆菌DH5α,经菌落PCR鉴定,每个样品挑选不少于3个阳性克隆交由上海生工生物科技有限公司测序,挑选3个克隆的测序结果保持一致的序列用于遗传多样性分析。
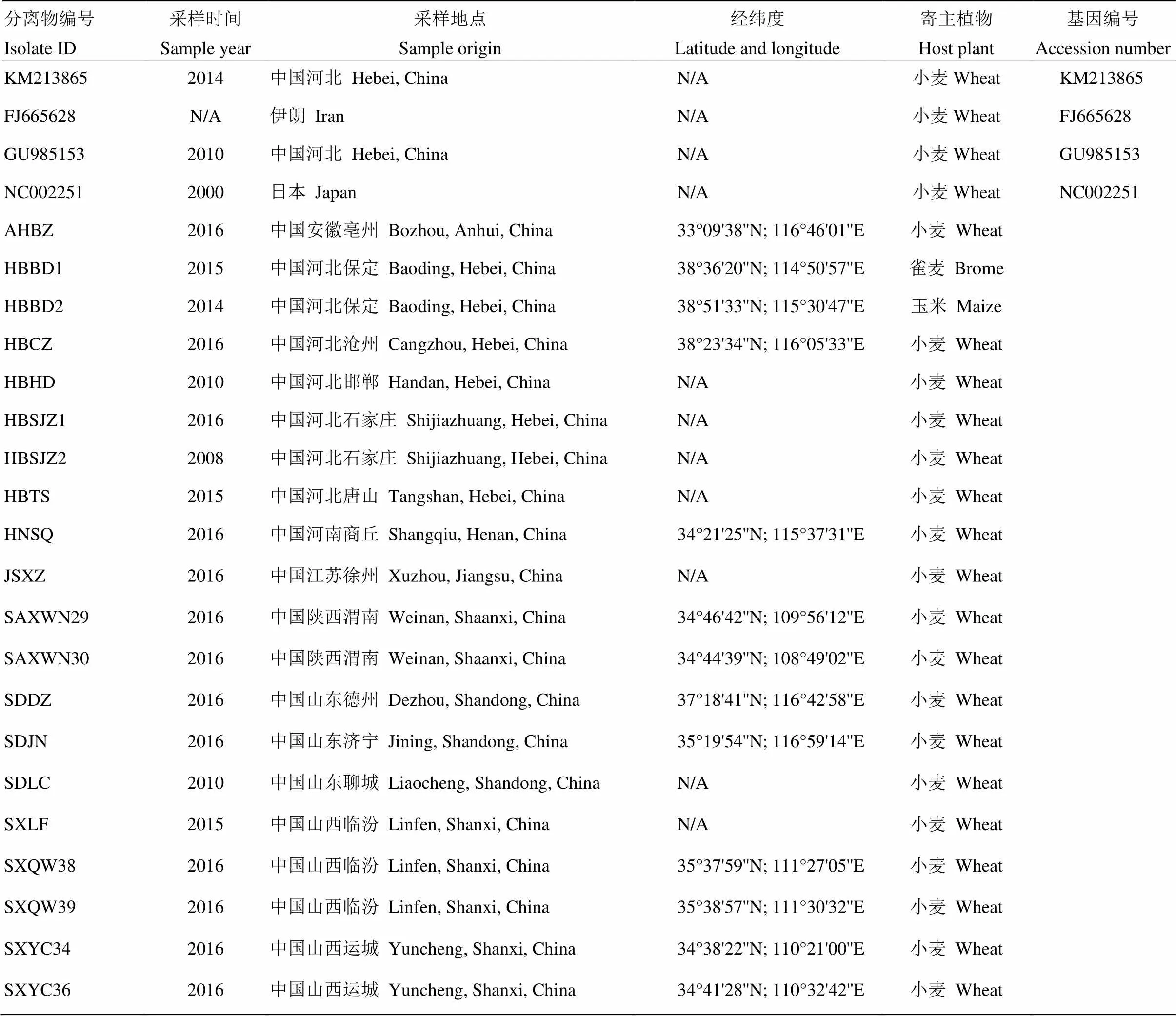
表1 大麦黄条点花叶病毒分离物信息

表2 大麦黄条点花叶病毒L、N基因引物
1.6 系统发育分析与遗传距离
选取大麦黄条点花叶病毒20个具有代表性的分离物(表1)及GenBank中河北分离物相应序列(GenBank登录号:KM213865),以GenBank中与大麦黄条点花叶病毒同属的、亲缘关系相近的北方禾谷花叶病毒相应序列(GenBank登录号:NC002251日本、GU985153河北)作为系统发育树的外族群,利用MEGA v6.0[21]软件ClustalW进行序列比对,删除引物及gap;采用最大似然法作图,自展1 000次,基于Tamura 3-parameter模型,构建系统发育树,不显示小于50%的自展值。使用泊松校正方法计算各分离物间及系统发育树各分组间的平均遗传距离。以L基因构建系统发育树,加入了GenBank中伊朗分离物的相应序列(GenBank登录号:FJ665628)。
1.7 遗传重组和选择压力分析
由MEGA软件进行基因序列比对,而后利用RDP v4.80程序包[22]进行重组分析。该程序包RDP[23]、GENECONV[24]、MaxChi[25]、Bootscan[26]、Chimaera[27]、SiScan[28]和3Seq[29]有4种以上软件分析结果支持重组,且p<1.0×10-6时,该分离物被认为存在“明确”重组,否则认为不存在重组或存在“潜在”重组。
利用PAML v4.9c软件包[30]中的codeml程序,使用位点模型对L和N基因进行选择压力分析。根据ω(dN/dS)值衡量选择压力,ω>1为正选择,ω<1为负选择,ω=1为中性选择[31-32]。设置参数Model=0,NSsites=0、1、2、3、7、8,对模型M0/M3、M1a/M2a和M7/M8进行似然比检验(LRT),评估合适的模型,通过PAML中的Bayesian方法计算发生正选择的氨基酸位点[33-34]。
1.8 遗传多样性和遗传分化分析
利用DnaSP v5[35]计算单倍型多样性(haplotype diversity,Hd)、核苷酸多样性(nucleotide diversity,π)等遗传变异统计量,并计算群体间的遗传分化系数(FST)和基因流(Nm)。根据FST值判断群体的分化程度,FST值在0—0.05表示群体间不存在分化,在0.05—0.15为中度分化,在0.15—0.25为高度分化,>0.25表明分化度极大。分别采用KS*、Z和Snn检验群体间的遗传分化显著水平。
2 结果
2.1 大麦黄条点花叶病毒的分布
大麦黄条点花叶病毒与北方禾谷花叶病毒同为细胞质弹状病毒属成员,二者亲缘关系相近,可在田间小麦上形成相似症状,而水稻黑条矮缩病毒也侵染小麦造成危害。应用一步法三重RT-PCR技术检测田间疑似病毒侵染的小麦病株,PCR产物电泳结果显示阳性对照扩增产物电泳条带清晰,BYSMV、RBSDV、NCMV基因扩增片段大小分别为254、354和509 bp,阴性对照未扩出任何条带,田间病株样品检测出3种病毒,且有混合侵染发生(图1)。
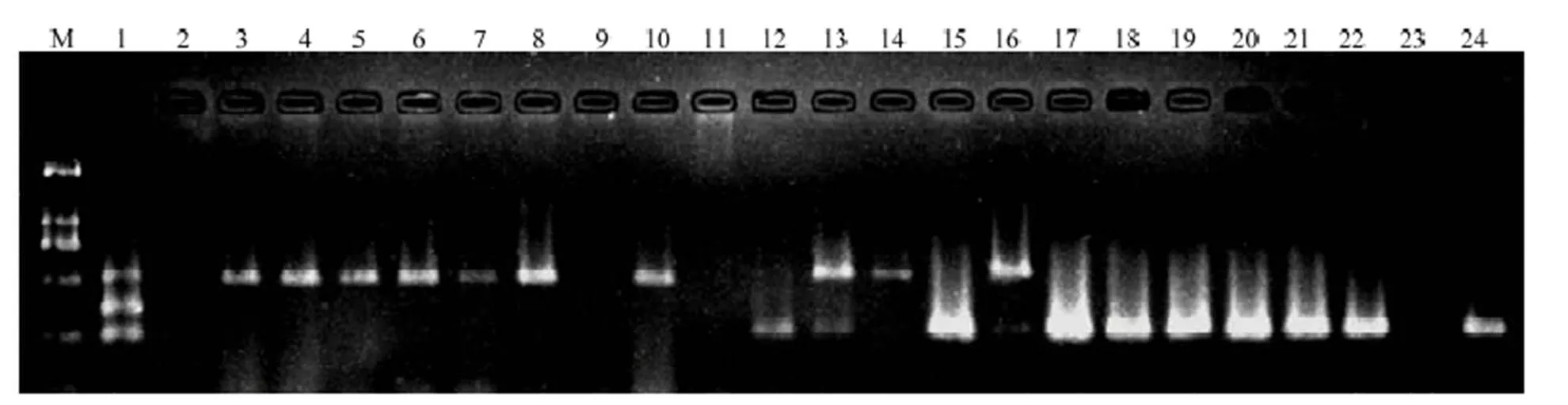
M:DL2000;1:阳性对照Positive control;2:阴性对照Negative control;3—24:田间样品Some of field samples
为探明BYSMV在中国北方小麦主产区的分布,共检测了北方小麦主产区7个省66个县/市/区的864份显典型病毒病症状的小麦、玉米和雀麦病株样品。检测结果表明,其中48个县/市/区采集的336份样品检测到BYSMV,平均检出率为38.89%。陕西的样品检出率最高,为85.71%,其次为河北的检出率63.05%,而江苏的检出率最低为13.18%。因此,BYSMV在调查的7个省份均有分布,且主要分布于陕西、河北、山西、山东,在河南及安徽北部亦有分布,在江苏徐州和邳州仅局部发生(表3)。

表3 不同地区标样中大麦黄条点花叶病毒、水稻黑条矮缩病毒和北方禾谷花叶病毒的检测结果
此外,36份样品中检测到RBSDV,157份样品检测到NCMV。河南样品中NCMV的检出率最高,为50.34%;其次为安徽,NCMV检出率为35.71%;山东、河北、陕西和山西的样品中NCMV检出率为7.28%—15.09%。因此,NCMV在除江苏以外的6省均有分布,且主要分布于河南及安徽北部;在采样最密集的河北,仅在北部唐山和南部邯郸两地发现。
2.2 系统发育分析
基于L基因片段构建的系统发育树可将22个BYSMV分离物划分为2个亚组(亚组I和II)。山西2个分离物SXQW39和SXYC34以及陕西分离物SAXWN30与伊朗分离物归为亚组II,其余17个分离物与河北分离物归为亚组I,2个亚组均与NCMV外族群明显划分开来(图2-a)。亚组I、亚组II分离物间平均遗传距离分别为0.0039、0.0062,2个亚组间遗传距离为0.0345(表4)。山西和陕西的分离物在2个亚组中均有分布,而其余5省的分离物则均划分在同一亚组中,说明山西、陕西地区BYSMV的遗传多样性水平较高。亚组I的基因型的分布范围较广,为优势基因型。系统发育树分组与寄主植物和采样时间无明显相关性。
基于N基因片段构建的系统发育树可将21个BYSMV分离物可以划分为2个亚组(亚组I和II)。其中山西3个分离物SXQW39、SXYC36、SXYC34和陕西分离物SAXWN30划为亚组II,其余16个分离物与河北分离物归为亚组I,2个亚组均与NCMV外族群明显划分开来(图2-b)。亚组I分离物间平均遗传距离为0.0053,亚组II为0.0049,2个亚组间遗传距离为0.0273(表4)。
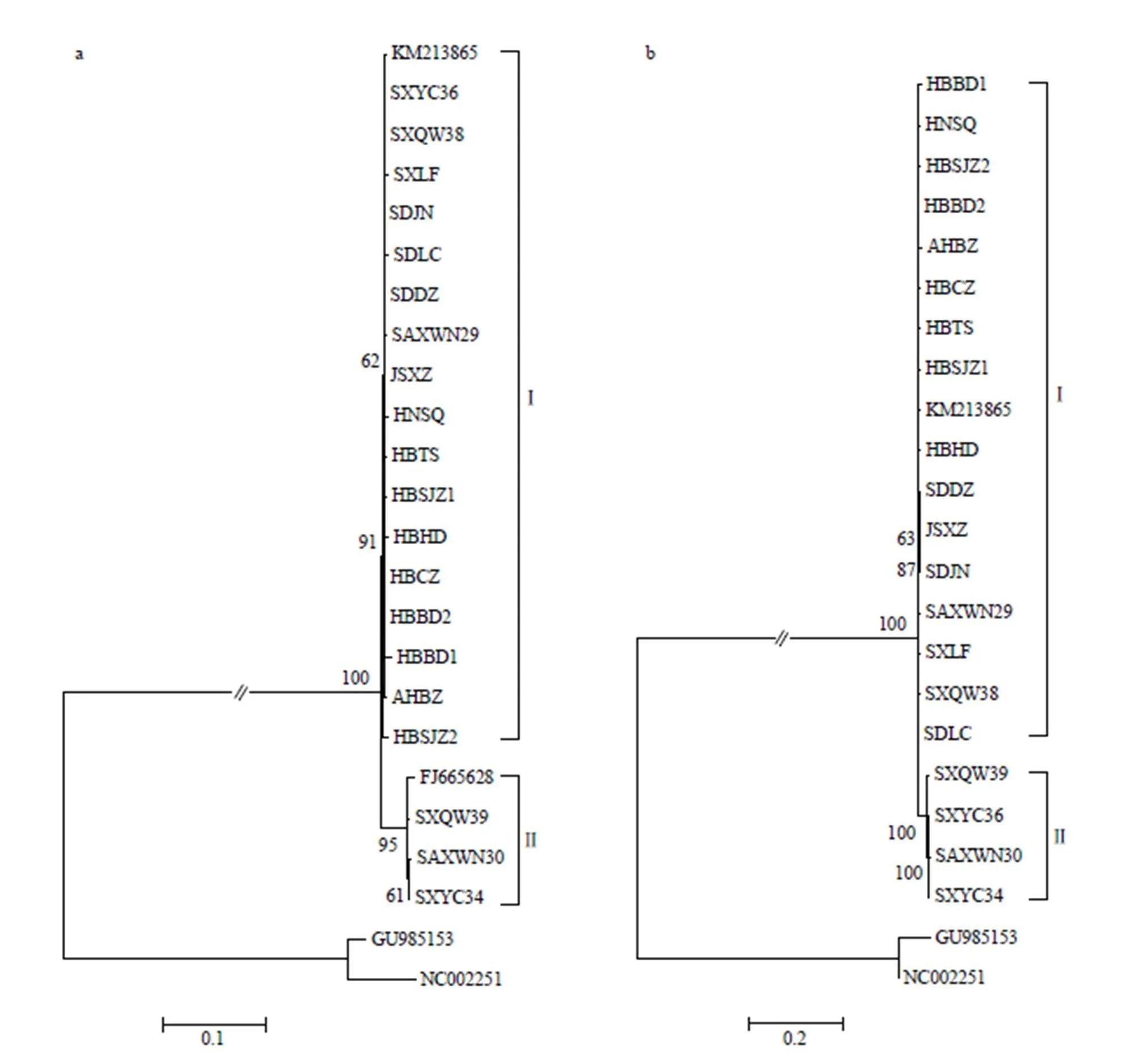
图2 基于大麦黄条点花叶病毒L(a)、N(b)基因片段的系统发生树(以北方禾谷花叶病毒为外群)
对比L和N基因的建树结果,发现N基因片段构建的树同样显示山西、陕西地区BYSMV群体的具有较高的遗传多样性水平,且亚组I的基因型为优势基因型;此外,编号为SXYC36的山西分离物在以L基因片段和N基因片段构建的系统发育树中分别属于亚组I和亚组II,推测该分离物的基因组可能发生了重组。
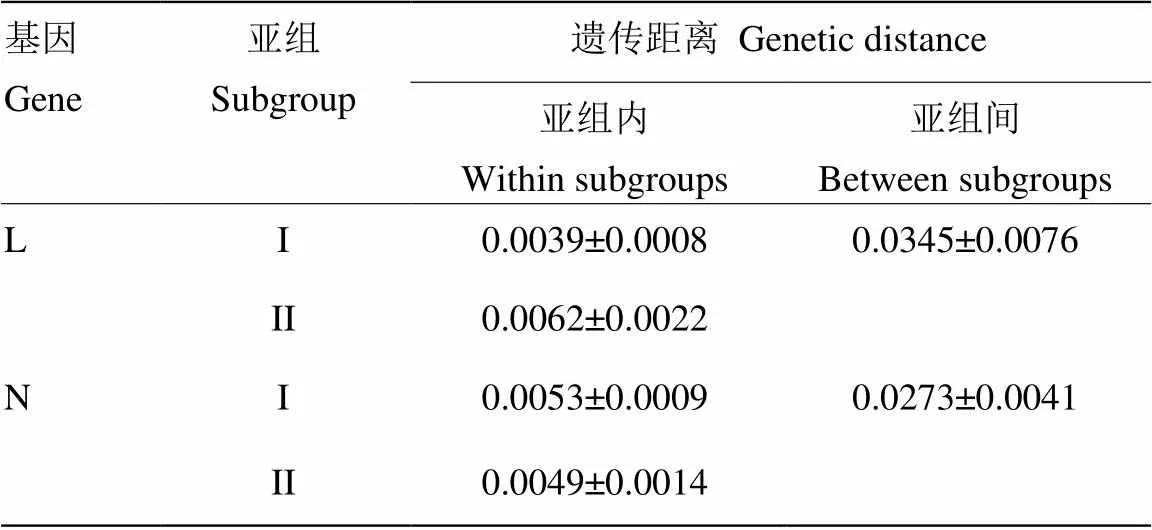
表4 大麦黄条点花叶病毒亚组内和亚组间的平均遗传距离
2.3 遗传重组及选择压力
RDP、GENECONV、MaxChi、Bootscan、Chimaera、SiScan和3Seq对L和N基因片段进行遗传重组分析,均未发现基因重组证据。
对L和N基因片段的选择压力分析表明,在M0(one ratio)模式下,亚组内和亚组间的ω(dN/dS)值的变异范围是0.02—0.19,远小于1,没有直接证据表明存在正选择(表5)。不同模型似然值lnL卡方检验结果表明M0与M3差异不显著,表明不同位点受到的选择压力没有差异;M1a(neutral)和M2a(selection)差异不显著,M2a未能检测到正选择位点;M7(beta)和M8(beta & ω)差异不显著,M8未能检测到正选择位点。因此,L和N基因片段均受到较强的净化选择的约束。
2.4 遗传多样性及遗传分化
遗传多样性分析表明,L基因和N基因变异均为碱基置换,其中L基因共有42个(7.07%)多态性位点(polymorphic sites),有34个多态性位点是密码子的第3碱基置换,此外有5个和3个多态性位点碱基置换分别是密码子的第2碱基和第1碱基。N基因共有76个(6.13%)多态性位点(表5),密码子第3碱基置换的有63个,密码子的第1碱基和第2碱基置换的分别有9个和4个。L基因和N基因均无插入/缺失突变。
22个分离物的L基因中共检测到16种单倍型,其中15种见于单个分离物,1种为7个分离物共有。21个分离物的N基因中共检测到20种单倍型,其中19种见于单个分离物,1种为2个分离物共有。L和N基因的Hd值均大于0.5、π值均高于0.005,表明中国BYSMV群体遗传多样性高(表5)。
根据地理来源,将河北、山东、河南、安徽、江苏的分离物划分为东部群体,山西、陕西和伊朗的分离物划分为西部群体,以L和N基因为分子标记分析BYSMV的分化程度,结果表明东部、西部群体的F值为0.32201和0.37326,均大于0.25(wright,1978),且、、3种统计检验显著(<0.05),表明东部和西部的BYSMV群体严重分化,而且差异显著(表6)。基因流(Nm)值分别为0.53和0.42,均小于1,说明有限的基因流是促使群体发生遗传分化的主要原因。
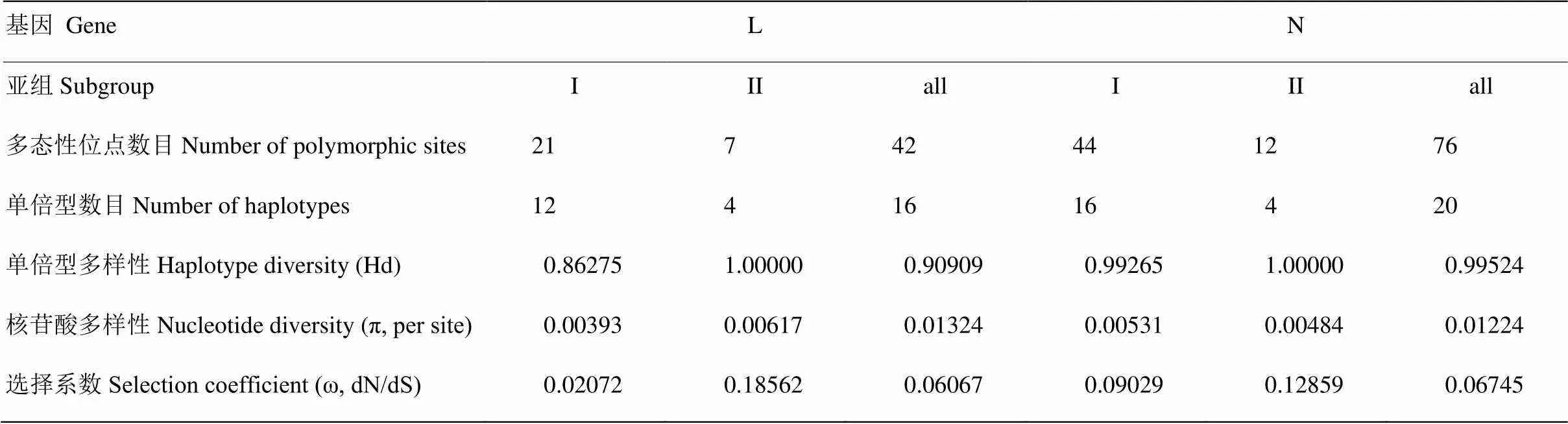
表5 基于L和N基因片段的遗传多样性分析

表6 大麦黄条点花叶病毒遗传分化分析
*表示排列检验重复1 000次得到的值在0.01—0.05,**表示0.001<<0.01,***表示<0.001
* means significant difference at 0.01<<0.05 level by permutation test with 1 000 replicates; ** means 0.001<<0.01; *** means<0.001
3 讨论
本研究通过对中国北方小麦主产区田间调查和病株样品的检测,发现大麦黄条点花叶病毒(BYSMV)在河北、山东、山西、陕西、河南北部等地已有广泛分布。分析不同时期样品检测结果表明,2010年BYSMV的检出率为38.89%,而2016年的检出率上升至48.89%,因此大麦黄条点花叶病毒田间危害有蔓延趋势。北方禾谷花叶病毒(NCMV)曾在河北等地造成严重危害[36],但近年来其流行危害减轻,田间调查和室内病毒检测也进一步证实NCMV的检出率降低,2010年在河北、河南、山东和山西4省疑似感染病毒的小麦中NCMV的检出率达91.3%[37],但到2016年降至18.08%,而且2015—2016年河北廊坊、保定、石家庄、衡水、邢台、沧州及山西运城等地样品中均未再检测到NCMV。BYSMV与NCMV的氨基酸相似性为35%—62%[11],且二者均由介体灰飞虱传播,但BYSMV与 NCMV的流行趋势则呈现出相反的结果,2种病毒的发生动态变化与环境、介体和寄主的相互关系值得深入研究。
基于BYSMV分离物L和N基因片段的系统发育分析表明,BYSMV分离物均可被分为2个亚组,分组与地理来源有一定的相关性;亚组I的基因型在中国北方分布范围广,在群体中所占比例高,为优势基因型;亚组II的基因型仅在山西、陕西分布,在基于L基因片段构建的系统发育树中该基因型伊朗分离物亲缘关系较近[12]。此外,研究显示有限的基因交流频率促使BYSMV种群有较高的遗传分化,因此,推测地理隔离是BYSMV种群分化的主要原因。目前关于BYSMV分离物基因序列的报道较少,BYSMV传入中国的来源和途径尚不清楚。
BYSMV是负单链RNA病毒,由于RNA病毒具有群体数量大、极高的复制速率、复合酶缺乏校对活性和基因组小等特点,基因组容易发生突变,从而增强病毒群体的适应能力[38]。中国小麦种植区域广泛,生态条件复杂,因此,BYSMV在局部地区极有可能暴发危害,及时监控BYSMV的流行及基因变异动态对于病害防控具有重要意义。
4 结论
大麦黄条点花叶病毒(BYSMV)在中国北方小麦主产区分布广泛,河北、山东、江苏、安徽、河南、陕西和山西等地均有不同程度的发生。系统发育分析可将BYSMV分离物分为2个亚组,分组与分离物的地理来源有一定的相关性。BYSMV群体具有丰富的遗传多样性,且东部、西部群体之间存在严重的遗传分化,有限的基因流是促使群体发生遗传分化的主要原因。
致谢:
在样品采集过程中得到江苏省农业科学院植物保护研究所程兆榜研究员,西北农林科技大学旱区作物逆境生物学国家重点实验室刘同先教授,河北省农林科学院植物保护研究所孔令晓研究员、纪莉景副研究员、闫冲,河北省第六人民医院崔利军主任,河南农业大学植物保护学院李洪连教授、孙炳剑副教授,山东省临沂市农业技术推广中心李宝庆等的大力支持。在此一并表示感谢!
[1] CONTI M. Vector relationships and other characteristics of(BYSMV)., 1980, 95(1): 83-92.
[2] 邸垫平, 张永亮, 张爱红, 闫冲, 杨菲, 路银贵, 田兰芝, 王献兵, 苗洪芹. 灰飞虱传播的一种小麦病毒病鉴定. 植物病理学报, 2016, 46(4): 453-460.
DI D P, ZHANG Y L, ZHANG A H, YAN C, YANG F, LU Y G, TIAN L Z, WANG X B, MIAO H Q. Identification of a virus transmitted by small brown planthopper in wheat., 2016, 46(4): 453-460. (in Chinese)
[3] DI D P, ZHANG Y L, YAN C, YAN T, ZHANG A H, YANG F, CAO X L, LI D W, LU Y G, WANG X B, MIAO H Q. First report ofon wheat in China., 2014, 98(10): 1450.
[4] CONTI M. Investigation on a bullet-shaped virus of cereals isolated in Italy from planthoppers., 1969, 66(3): 275-279.
[5] GREBER R S. Maize sterile stunt- a delphacid transmitted rhabdovirus affecting some maize genotypes in Australia., 1982, 33(1): 13-23.
[6] Lockhart B E L, El-Maataoui M, Carroll T W, Lennon A M, Zaske S K. Identification ofin Morocco and its field detection by enzyme immune assay., 1986, 70(12): 1113-1117.
[7] Izadpanah K, Ebrahim-Nesbat F, Afsharifar A R.as the cause of a major disease of wheat and millet in Iran., 1991, 131(4): 290-296.
[8] Makkouk K M, Bertschinger L, Conti M, Bolay N, Dusunceli F.naturally infects cereal crops in the Anatolian Plateau of Turkey., 1996, 144(7/8): 413-415.
[9] MAKKOUK K, KUMARI S G, GHULAM W, ATTAR N. First record ofaffecting wheat Summer- Nurseries in Syria., 2004, 88(1): 83.
[10] DUMÓN A D, Argüello-Caro E B, Alemandri M V, Bainotti C, Mattio M F, Rodríguez S M, del Vas M, Truol G. Identification and biological characterization of(BYSMV): A new wheat disease in Argentina., 2011, 36(6): 374-382.
[11] Yan T, Zhu J R, Di D P, Gao Q, Zhang Y L, Zhang A H, Yan C, Miao H Q, Wang X B. Characterization of the complete genome ofreveals a nested gene encoding a small hydrophobic protein., 2015, 478: 112-122.
[12] ALMASI R, AFSHARIFAR A, NIAZI A, PAKDEL A, IZADPANAH K. Analysis of the complete nucleotide sequence of the polymerase gene of– Iranian isolate., 2010, 158(5): 351-356.
[13] ROOSSINCK M J. Mechanisms of plant virus evolution., 1997, 35: 191-209.
[14] DRAKE J W, HOLLAND J J. Mutation rates among RNA viruses., 1999, 96(24): 13910-13913.
[15] WEI T Y, YANG J G, LIAO F R, GAO F L, LU L M, ZHANG X T, LI F, WU Z J, LIN Q Y, XIE L H, LIN H X. Genetic diversity and population structure ofin China., 2009, 90(4): 1025-1034.
[16] WU B, BLANCHARD-LETORT A, LIU Y, ZHOU G, WANG X, ELENA S F. Dynamics of molecular evolution and phylogeography of-PAV., 2011, 6(2): e16896.
[17] YIN X, ZHENG F Q, TANG W, ZHU Q Q, LI X D, ZHANG G M, LIU H T, LIU B S. Genetic structure ofpopulations in China., 2013, 158(12): 2505-2515.
[18] Higgins C M, Chang W L, Khan S, Tang J, Elliott C, Dietzgen R G. Diversity and evolutionary history ofin Australia and New Zealand., 2016, 161(2): 269-277.
[19] Klerks M M, Lindner J L, Vaškova D, Špak J, Thompson J R, Jelkmann W, Schoen C D. Detection and tentative grouping ofisolates., 2004, 110: 45-52.
[20] Pappi P G, Maliogka V I, Amoutzias G D, Katis N I.Genetic variation offrom annual and perennial plant hosts., 2016, 161(3): 631-639.
[21] Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6:Molecular evolutionary genetics analysis version 6.0., 2013, 30(12): 2725-2729.
[22] Martin D P, MURRELL B, golden M, KHOOSAL A, MUHIRE b. RDP4: Detection and analysis of recombination patterns in virus genomes., 2015, 1(1): vev003.
[23] MARTIN D, RYBICKI E. RDP: detection of recombination amongst aligned sequences., 2000, 16(6): 562-563.
[24] PADIDAM M, SAWYER S, FAUQUET C M. Possible emergence of new geminiviruses by frequent recombination., 1999, 265(2): 218-225.
[25] SMITH J M. Analyzing the mosaic structure of genes., 1992, 34(2): 126-129.
[26] MARTIN D P, POSADA D, CRANDALL K A, WILLIAMSON C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints., 2005, 21(1): 98-102.
[27] POSADA D, CRANDALL K A. Evaluation of methods for detecting recombination from DNA sequences: computer simulations., 2001, 98(24): 13757-13762.
[28] GIBBS M J, ARMSTRONG J S, GIBBS A J. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences., 2000, 16(7): 573-582.
[29] BONI M F, POSADA D, FELDMAN M W. An exact nonparametric method for inferring mosaic structure in sequence triplets., 2007, 176(2): 1035-1047.
[30] Yang Z H. PAML 4: a program package for phylogenetic analysis by maximum likelihood., 2007, 24(8): 1586-1591.
[31] SWANSON W J, VACQUIER V D. The rapid evolution of reproductive proteins., 2002, 3(2): 137-144.
[32] SWANSON W J, NIELSEN R, YANG Q F. Pervasive adaptive evolution in mammalian fertilization proteins., 2003, 20(1): 18-20.
[33] Yang Z H, Nielsen R, Goldman N, Pedersen A M K. Codon-substitution models for heterogeneous selection pressure at amino acid sites., 2000, 155(1): 431-449.
[34] Yang Z H, Wong W S W, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection., 2005, 22(4): 1107-1118.
[35] Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data., 2009, 25(11): 1451-1452.
[36] 裘维蕃, 杨莉, 梅汝鸿, 林志亮, 蔡祝南. 小麦丛矮病研究之一分布、症状及损失. 植物保护学报, 1979, 6(1): 11-16.
QIU W F, YANG L, MEI R H, LIN Z L, CAI Z N. Studies on a rosette disease of wheat I distribution, symptoms and losses., 1979, 6(1): 11-16. (in Chinese)
[37] 段西飞, 邸垫平, 张爱红, 苗洪芹. 中国北方四省小麦丛矮病病原鉴定. 植物病理学报, 2013, 43(1): 91-94.
DUAN X F, DI D P, ZHANG A H, MIAO H Q. Molecular detection and identification of the pathogen causing wheat rosette stunt disease in the north of China., 2013, 43(1): 91-94. (in Chinese)
[38] MOYA A, HOLMES E C, GONZÁLEZ-CANDELAS F. The population genetics and evolutionary epidemiology of RNA viruses., 2004, 2(4): 279-288.
(责任编辑 岳梅)
Distribution and genetic diversity ofin northern China
YANG Fei, ZHANG AiHong, MENG FanSi, HUO LiangZhan, LI XiWang, DI DianPing,MIAO HongQin
(Plant Protection Institute of Hebei Academy of Agricultural and Forestry Sciences/IPM Center of Hebei Province/Key Laboratory of Integrated Pest Management on Crops in Northern Region of North China, Ministry of Agriculture, Baoding 071000, Hebei)
【Objective】The objective of this study is to clarify the distributionand genetic diversity of(BYSMV) in major wheat production areas in northern China, andto provide a theoretical basis for the early warning, prevention and control of epidemic diseases.【Method】During 2008-2016, about 864 suspected virus-infected samples were collected from 66 districts in Hebei, Shandong, Jiangsu, Anhui, Henan, Shaanxi and Shanxi provinces, all the three viruses including BYSMV,(RBSDV) and(NCMV) were identified using one-step multiplex RT-PCR. L and N gene fragments of BYSMV were obtained by RT-PCR amplification and cloned, and then determined by nucleotide sequence analysis. The sequences were analyzed by softwares of MEGA, DnaSP, and PAML to elucidate the phylogenesis and genetic diversity of BYSMV isolates. 【Result】A total of 336 samples collected from 48 districts in 7 provinces were detected with BYSMV and the detection rate was 38.89%. The virus was mainly distributed in Shaanxi, Hebei, Shanxi and Shandong. In addition, it was also distributed in Henan and northern Anhui. Xuzhou and Pizhou in Jiangsu Province were only localized. Phylogenetic analysis showed that the population could be divided into two subgroups (I and II) based on fragments of L and N genes. The isolates in subgroup I were derived from all 7 provinces, but isolates in subgroup II were only from Shaanxi and Shanxi provinces. It was indicated that Iran isolate was related to the subgroup II isolates based on the phylogenesis of L gene sequence. The clustering of the isolates was related to their geographical origins, andnot to the host plants or sampling dates.Genetic analysis by using 7 softwares of the RDP package showed that there was no evidence supported for therecombination. Selection stress analyses showed that the ω (dN/dS) values varied from 0.02 to 0.19 which were far less than 1 within or between subgroups. It was indicated that the population was undergoing purifying selection. Haplotype diversity (Hd) values (0.90909 and 0.99524) of L and N gene fragments were greater than 0.5 and nucleotide diversity (π) values (0.01324 and 0.01224) were higher than 0.005, indicating that there was a high level of genetic diversity in the population of BYSMV in China. genetic differentiation based on L and N gene fragments showed thatthe fixation indicesF(0.32201 and 0.37326) of eastern and western subpopulations were greater than 0.25. The difference of statistical test was significant, which indicated that the BYSMV population in eastern and western regions was seriously differentiated.The Nm values (0.53 and 0.42) were less than 1, which indicated that the limited gene flow was the main reason of genetic differentiation.【Conclusion】BYSMV is widely distributed in wheat producing areas of northern China, and occurred in Hebei, Shandong, Jiangsu, Anhui, Henan, Shaanxi and Shanxi provinces on different levels. The population of BYSMV had a high level of genetic diversity in China, and there is a severe genetic differentiation between the eastern and western subpopulations.
(BYSMV); phylogenesis; genetic diversity; selection pressure; genetic differentiation
2017-07-20;
2017-09-06
国家“973”计划(2014CB138400)、植物病虫害生物学国家重点实验室开放基金(SKLOF201614)
杨菲,E-mail:feiyang_987@163.com。
邸垫平,E-mail:chmrdv@163.com。通信作者苗洪芹,E-mail:miao5058345@163.com
