一种高效表达碱性蛋白酶的新型启动子的筛选及研究
2018-01-31陈坤袁飞燕柴昊男刘焕王兴吉张会图路福平
陈坤 袁飞燕 柴昊男 刘焕 王兴吉 张会图 路福平
(1. 工业发酵微生物教育部重点实验室 天津科技大学生物工程学院,天津 300457;2. 山东隆科特酶制剂有限公司,沂水 276400)
蛋白酶是一类能够催化水解肽键的酶,在食品、环境、医药等各类领域具有重要的应用价值[1-2],主导全球工业酶市场[3]。碱性蛋白酶(Alkaline protease)是一类最适pH值偏碱性的蛋白酶,在食品、洗涤以及皮革等行业有着广泛的用途[4-5]。碱性蛋白酶强的水解、耐热及耐碱能力[6],使其一直以来备受人类研究的关注。
由于碱性蛋白酶的广泛用途以及产酶效率不高等原因,一直以来市场都处于供不应求的现状[7]。目前,国内外对于微生物碱性蛋白酶的研究主要侧重于菌种选育、酶转译及调控机制的研究等方面[8-9],而对于提高生产菌产酶能力和酶的性质的研究较少。近年来,越来越多的研究者开始将注意力转向通过生物技术和蛋白质手段构建碱性蛋白酶高产工程菌等方面[10]。
枯草芽孢杆菌由于其无致病性、安全、无明显密码子偏嗜性、代谢产物分离纯化简便等优点[11-12],被广泛作为酶与抗生素等的表达宿主应用于工业生产。另一方面,枯草芽孢杆菌高效表达元件严重匮乏、遗传转化效率低和分泌系统较弱等问题[13-14],同时也限制了其应用价值。
在微生物遗传系统中,启动子具有十分重要的作用,选用强启动子介导目的基因的表达是提高基因异源表达非常有效的方法[15]。从地衣芽孢杆菌中筛选获得一种新型的强组成型启动子,以两种碱性蛋白酶基因为报告基因,研究其在枯草芽孢杆菌WB600中的表达强度,以期构建碱性蛋白酶高效表达系统,并为枯草芽孢杆菌异源基因的表达提供新型调控元件。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 枯草芽孢杆菌(B.subtilis)WB600、地衣芽孢杆菌(B.licheniformis)11965和枯草芽孢杆菌表达载体pUBA110均为本实验室保藏。1.1.2 试剂和培养基 DNA聚合酶、限制性内切酶、T4 DNA Ligase均购自TaKaRa公司;GeneRuler 1 kb DNA Ladder、Pierce Unstained Protein MW Marker 购自Thermo公司;质粒提取试剂盒、DNA切胶回收试剂盒购自OMEGA公司;福林酚、酪蛋白底物、三氯乙酸等购自Sangon Biotech(Shanghai)公司。
种子培养基(肉汤培养基):牛肉膏0.5%,蛋白胨1%,氯化钠0.5%,初始pH值为7.2-7.4;发酵产酶培养基:玉米粉6.4%,豆饼粉4%,磷酸氢二钠0.4%,磷酸二氢钾0.03%,高温淀粉酶0.07%(防止糊化)。
枯草芽孢杆菌感受态制备培养基:(1)SP-A Salts Solution :(NH4)2SO40.4%,K2HPO4·3H2O 2.8%,KH2PO41.2%,Trisodium Citrate Dihydrate 0.2% ;(2)SP-B Salts Solution :MgSO4·7H2O 0.04% ;(3)100×CAYE Solution :Casamino acid 2%,Yeast Extract 10% ;(4)SPI Medium(20 mL):SP-A Salts Solution 9.8 mL,SP-B Salts Solution 9.8 mL,50% Glucose 200 μL,100×CAYE 200 μL ;(5)SPII Medium(6 mL):SPI Medium 5.88 mL,50 mmol/L CaCl260 μL,250 mmol/L MgCl260 μL ;(6)100×EGTA Solution:10 mmol/L EGTA溶液,溶解时加入少量NaOH至pH 8.0。
1.2 方法
1.2.1 地衣芽孢杆菌启动子筛选 由本实验室菌种保藏库中接种地衣芽孢杆菌11965于无抗LB平板上,37℃培养12 h,挑取单菌落于新的LB平板三区划线进行活化,12 h后挑取单菌落于50 mL种子培养基中,220 r/min过夜培养,分别接取2 mL种子培养液于3个装有100 mL发酵培养基的500 mL挡板瓶中,37℃,220 r/min振荡培养,每隔8 h进行取样,离心收集上清液,进行SDS-PAGE蛋白电泳分析,切取最亮的条带进行蛋白质谱检测。
1.2.2 启动子及信号肽片段的克隆 蛋白质谱结果显示该酶为地衣芽孢杆菌几丁质酶(Chitinase),由NCBI数据库获取其基因序列,通过在线软件Promoter 2.0 Prediction Server(http://www.cbs.dtu.dk/services/Promoter/)和SignalP 4.1 Server(http://www.cbs.dtu.dk/services/SignalP/)分析其启动子和信号肽。Degering等[16]的研究表明源于地衣芽胞杆菌几丁质酶的信号肽dBli00338使蛋白酶subtilisin BPN'分泌表达量提高了6-7倍,因此选用dBli00338代替报告基因自身信号肽。通过引物pChi-F和SP-R(表1),以地衣芽孢杆菌11965基因组为模板,PCR扩增几丁质酶启动子和信号肽片段。PCR反应体系为:无菌双蒸水 33.8 μL,10×Pyrobest Buffer 5 μL,dNTP Mixture(2.5 mmol/L)5 μL,上游引物和下游引物各2 μL,基因组模板2 μL,Pyrobest DNA聚合酶0.2 μL。PCR反应条件为:94℃预变性5 min;94℃变性30 s,54℃退火30 s,72℃延伸30 s,共30个循环;72℃延伸10 min。0.8% 的琼脂糖凝胶电泳检测,切胶回收PCR产物。

表1 引物及核苷酸序列
选用启动子pShuttle-09和pyxiE为对照,验证pChi的表达强度。启动子pShuttle-09和pyxiE分别通过引物pS-F/pS-R和pY-F/pY-R(表1)经PCR扩增获得,并与引物SP-F/SP-R扩增的信号肽dBli00338片段通过重叠PCR技术进行连接。
1.2.3 两种碱性蛋白酶成熟肽基因的克隆 报告基因所用的两种碱性蛋白酶基因分别为克劳氏芽孢杆菌碱性蛋白酶基因alk(GenBank Sequence ID:FJ940727.1)和地衣芽孢杆菌碱性蛋白酶基因apr(GenBank Sequence ID:AY590140.1)。根据编码基因序列,运用在线分析软件SignalP 4.1 Server分析得到两种蛋白酶基因的成熟肽序列,并设计PCR引物alk-F/alk-R和apr-F/apr-R(表1)。
1.2.4 表达载体的构建 PCR产物切胶回收后获得的启动子-信号肽片段与报告基因成熟肽片段分别使用Bam HI酶切后连接,连接产物纯化后使用Sac II和Xho I双酶切并切胶回收,通过T4 DNA连接酶克隆到pUBA110表达载体上,连接反应体系为(10 μL):pUBA110载体片段3 μL,启动子-信号肽-报告基因片段 5 μL,10×T4 DNA Buffer 1 μL,T4 DNA连接酶1 μL,16℃连接6 h,连接产物通过化学转化转入枯草芽孢杆菌WB600感受态细胞。
1.2.5 枯草芽孢杆菌WB600感受态的制备及转化
(1)挑取新活化的枯草芽孢杆菌WB600单菌落于5 mL LB液体培养基中,37℃,220 r/min,过夜培养;(2)取100 μL培养液转接至5 mL SPI培养基中,37℃,220 r/min培养至对数生长末期OD600=1.2(约3-4 h);(3)取200 μL生长至对数期末的培养液至2 mL SPII培养基中,37℃,100 r/min 培养 1.5 h;(4)在上述SPII培养基的菌体中加入20 μL 10 mmol/L EGTA,37℃,100 r/min培养10 min;(5)加入连接产物,37℃,100 r/min培养30 min;(6)调节转速至220 r/min,继续培养1.5 h,取菌液涂布于含有50 μg/mL卡那霉素(Kan)的LB筛选平板,37℃培养12 h,筛选阳性转化子进行验证。
将构建的重组质粒分别命名为pCalk110、pCapr110、pSalk110、pSapr110、pYalk110 和pYapr110,所对应的重组菌命名为C1、C2、S1、S2、Y1和 Y2。
1.2.6 蛋白酶粗酶液的制备及分析 将新鲜平板上的6株菌的单菌落分别接入50 mL Kan抗性种子培养基中,37℃、220 r/min振荡培养12 h,分别检测菌株种子液的OD600,以相同的接种量转接于含有Kan抗性的装有100 mL发酵培养基的挡板瓶中,每株菌做3个平行,于37℃、220 r/min发酵培养,每隔6 h收集发酵液,4℃、12 000 r/min离心取上清,用于SDS-PAGE分析和酶活测定。SDS-PAGE分析取用20 μL发酵上清液与5 μL 5×SDS-PAGE Loading Buffer混合均匀,沸水浴10 min后上样10 μL。
碱性蛋白酶酶活测定参照GB/T 23527-2009[17]附录B福林酚法进行,1个酶活力单位(U/mL)定义为1 mL酶液在40℃、pH10.5条件下反应1 min水解酪蛋白产生1 μg酪氨酸所需要的酶量。
2 结果
2.1 地衣芽孢杆菌启动子筛选
对地衣芽孢杆菌11965进行发酵培养,分别取24 h,32 h,40 h,48 h的发酵上清液,使用12% 的蛋白分离胶检测,如图1所示,分子量大小为60 kD左右的胞外蛋白表达量最高。切取48 h该酶的蛋白电泳条带进行蛋白质谱检测,如图2为对该蛋白多肽链打断后不同出峰时间的元素丰度。对各出峰时间肽链进行蛋白质二级质谱,结果如表2,通过NCBI数据库进行比对,初步确定该酶为地衣芽孢杆菌几丁质酶(Chitinase)。
2.2 重组表达载体的构建
含有不同启动子和报告基因的重组表达载体构建过程见图3。通过PCR扩增及DNA切胶回收,成功获得5条基因片段,分别为399 bp的pChidBli0338,314 bp的pShuttle-09-dBli0338,248 bp的pyxiE-dBli0338,1 062 bp的克劳氏碱性蛋白酶基因alk成熟肽和1 053 bp的地衣碱性蛋白酶基因apr成熟肽,以及通过Sac II-XhoI双酶切并切胶回收获得的3 729 bp的pUBA110载体片段(图4),大小均与预期相符。重组载体连接产物转化进入WB600感受态,通过Kan抗性平板筛选,分别挑取转化子提取质粒DNA进行验证并由北京华大基因科技有限公司测序,测序分析与实验结果完全一致,证明报告基因和启动子-信号肽片段成功克隆到载体pUBA110上,重组表达载体pCalk110、pCapr110、pSalk110、pSapr110、pYalk110和pYapr110及所对应的重组菌C1、C2、S1、S2、Y1和Y2构建成功。

图1 地衣芽孢杆菌11965发酵上清蛋白电泳

图2 断裂的目的蛋白不同出峰时间的元素丰度

表2 目的蛋白各肽段的总二级质谱结果

图3 含有不同启动子和报告基因的重组表达载体的构建
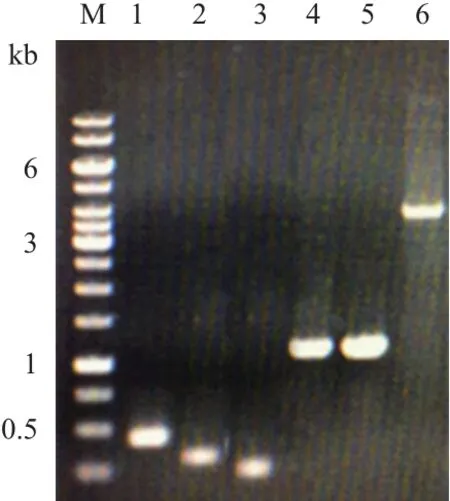
图4 重组载体各片段的PCR扩增及pUBA110的酶切回收
2.3 发酵上清液的SDS-PAGE检测
分别取C1、S1、Y1、C2、S2和Y2六株菌48 h的发酵上清液通过1.5 mL,10 K的超滤柱浓缩后进行蛋白电泳检测。SDS-PAGE结果(图5)显示,6株菌均有一条清晰的条带,且与目标蛋白分子量大小38.8 kD和31.3 kD相一致,表明在3种启动子的调控下,两种碱性蛋白酶报告基因均成功表达。
2.4 不同启动子对两种碱性蛋白酶基因表达强度的影响
测定不同时间发酵上清液中碱性蛋白酶的酶活,每组实验作3个平行,通过SPSS(Statistical Product and Service Solutions)软件对实验结果进行统计学分析并检测其显著性,实验结果由x-±s表示。如图6,发酵培养48 h时,各重组菌发酵上清液中碱性蛋白酶活性达到最高。筛选得到的启动子pChi对两种报告基因的启动强度明显高于另外两个启动子,发酵48 h时,pChi、pShuttle-09、pyxiE表达克劳氏碱性蛋白酶的活性分别为(8027.55±230.84)U/mL、(7124.62±295.48)U/mL、(5268.28±196.49)U/mL(P= 5.49×10-4< 0.01),表达地衣碱性蛋白酶的活性分别为(3545.42±146.97)U/mL、(2638.91±106.72)U/mL、(1472.78±89.65)U/mL(P= 6.74×10-6< 0.01)。结果表明,pChi的启动强度是pShuttle-09的1.25倍,pyxiE的2倍,该结果与蛋白电泳初步检测的结果相一致。
2.5 启动子pChi结构预测与分析
通过在线分析软件BPROM对筛选所获得启动子pChi进行结构预测和分析,结果如图7。pChi具有原核启动子典型的保守序列-35区(TTGTCT)和-10区(TATTAG),被σA因子识别,间隔为16 bp。转录起始位点(+1)与-10区之间的间隔为6 bp,核糖体结合位点(SD,AGGAGA)距离几丁质酶基因的起始密码子(GTG)8 bp。

图5 重组菌的蛋白电泳检测
3 讨论
Yang等[18]利用启动子诱捕技术由地衣芽孢杆菌中筛选的启动子pShuttle-09表达强度达到P43的8倍,pShuttle-09为杂合启动子,含有两对典型的保守序列,均被σA因子识别,分别为相距18 bp的-35区(TTGACG)和-10区(TATTAT),相距19 bp的-35区(CTGAAA)和-10区(TATAAT)。SD(GAGAGG)距离基因luxS的起始密码子(ATG)4 bp。Zhang等[19]由枯草芽孢杆菌筛选获得的启动子pyxiE表达强度也高于P43,pyxiE含有间隔为20 bp的-35区(TTGACA)和-10区(AATAAA),转录起始位点(+1)与-10区间隔为7 bp,典型的SD序列(AGGCGG)距离基因yxiE的起始密码子(ATG)7 bp。

图6 六株重组菌的产酶曲线

图7 启动子pChi结构分析
启动子的启动强度主要取决于启动子的结构,一般来说,两保守区序列-35区和-10区间隔为17 bp或18 bp时启动活性最强,而RBS结合位点与起始密码子之间的距离也是决定启动子强度的关键因 素,通 常 为 10 bp[20]。pChi、pShuttle-09、pyxiE三个启动子的保守序列均被σA因子识别,pChi和pShuttle-09两保守区之间间隔为16 bp和18 bp,但pyxiE为20 bp,这可能是造成pyxiE的启动活性远低于其它两种启动子的主要原因。而pChi和pShuttle-09中SD与起始密码子分别相距8 bp、4 bp,这可能是pChi表达活性高于pShuttle-09的原因之一。
4 结论
由地衣芽孢杆菌基因组中筛选获得一种新型组成型启动子,以克劳氏芽孢杆菌碱性蛋白酶和地衣芽孢杆菌碱性蛋白酶基因为报告基因,对其表达活性进行研究。结果显示,启动子pChi的表达强度是pShuttle-09的1.25倍,pyxiE的2倍,从而为介导枯草芽孢杆菌表达系统中异源基因的高效表达奠定基础。另一方面,构建了碱性蛋白酶高效表达工程菌,实现了碱性蛋白酶的高效分泌表达,推动碱性蛋白酶的基因工程改造进程以及工业化生产。
[1] Sharma KM, Kumar R, Panwar S, et al. Microbial alkaline proteases:Optimization of production parameters and their properties[J]. Journal of Genetic Engineering and Biotechnology,2017, 15(1):115-126.
[2] Rani K, Rana R, Datt S. Review on latest overview of protease[J].International Journal of Current Life Sciences, 2012, 2(1):12-18.
[3] Raval VH, Pillai S, Rawal CM, et al. Biochemical and structural characterization of a detergent-stable serine alkaline protease from seawater haloalkaliphilic bacteria[J]. Process Biochemistry,2014, 49(6):955-962.
[4] Amit G, Babu J, Abin M, et al. Biosynthesis and properties of an extracellular thermostable serine alkaline protease from Virgibacillus pantothenticus[J]. World Journal of Microbiology and Biotechnology, 2008, 24(2):237-243.
[5] Jose D, Manjusha K, Jose S, et al. Purification and characterization of highly active LasB protease from Pseudomonas aeruginosa MCCB 123[J]. Indian Journal of Experimental Biology, 2017, 55(5):303-310.
[6] 邓菊云. 微生物碱性蛋白酶研究进展[J]. 现代食品科技,2008, 24(3):293-296.
[7] Sharma KM, Kumar R, Vats S, et al. Production, partial purification and characterization of alkaline protease from Bacillus aryabhattai K3[J]. Chinese Journal of Applied and Environmental Biology,2014, 38(2):290-298.
[8] Jaouadi B, Ellouz-Chaabouni S, Rhimi M, et al. Biochemical and molecular characterization of a detergent-stable alkaline protease from Bacillus pumilus CBS with high catalytic efficiency[J].Biochimie, 2008, 90(9):1291-1305.
[9] Abidi F, Limam F, Marzouki MN. Purification and characterization of an alkaline protease prot1 from Botrysis cinerea[J]. Applied Biochemistry and Biotechnology, 2007, 141(2-3):361-376.
[10] Suseela L, Anjali CH, Muralidhar P. Enhanced production of alkaline protease by Aspergillus niger DEF 1 isolated from dairy form effluent and determination of its fibrinolytic ability[J].African Journal of Microbiology Research, 2017, 11(11):440-449.
[11] Liu L, Liu YF, Shin HD, et al. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology[J]. Applied Microbiology and Biotechnology, 2013, 97(14):6113-6127.
[12] Dong HN, Zhang DW. Current development in genetic engineering strategies of Bacillus species[J]. Microbial Cell Factories, 2014,13(1):63-73.
[13] Goosens VJ, Otto A, Glasner C, et al. Novel twin-arginine translocation pathway-dependent phenotypes of Bacillus subtilis unveiled by quantitative proteomics[J]. Journal of Proteome Research, 2012, 12(2):796-807.
[14] 余小霞, 田健, 刘晓青, 等. 枯草芽孢杆菌表达系统及其启动子研究进展[J]. 生物技术通报, 2015, 31(2):35-44.
[15] Phan TT, Nguyen HD, Schumann W. Development of a strong intracellular expression system for Bacillus subtilis by optimizing promoter elements[J]. Journal of Biotechnology, 2012, 157(1):167-172.
[16] Degering C, Eggert T, Puls M, et al. Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides[J]. Applied and Environmental Microbiology, 2010, 76(19):6370-6376.
[17] 中华人民共和国国家质量监督检验检疫总局, 中国国家标准化管理委员会. GB/T 23527-2009 蛋白酶制剂[S]. 北京:中国标准出版社, 2009.
[18] Yang MM, Zhang WW, Ji SY, et al. Generation of an artificial double promoter for protein expression in Bacillus subtilis through a promoter trap system[J]. PLoS One, 2013, 8(2):e56321.
[19] Zhang AL, Liu H, Yang MM, et al. Assay and characterization of a strong promoter element from B. subtilis[J]. Biochemical and Biophysical Research Communications, 2007, 354(1):90-95.
[20] Song YF, Nikoloff JM, Zhang DW. Improving protein production on the level of regulation of both expression and secretion pathways in Bacillus subtilis[J]. Journal of Microbiology and Biotechnology,2015, 25(7):963-977.
