同一家系相同CLCN5突变不同临床表型2例病例报告
2018-01-23张宏文刘晓宇肖慧捷
张宏文 王 芳 刘晓宇 肖慧捷 姚 勇
1 病例资料
例1 患儿男,10.9岁,因“发现蛋白尿5年余、多饮多尿4年”于2015年8月14日入住北京大学第一医院(我院)儿科肾脏病房。
患儿系G1P1,出生史无特殊记载。5年前体检时发现蛋白尿(78 mg·kg-1),不伴水肿、少尿,血白蛋白和胆固醇在正常值范围。外院肾脏病理检查:免疫荧光均阴性;光镜下可见13个肾小球,足细胞肿胀,系膜细胞和基质轻度增生,节段肾小球基底膜增粗,肾小管萎缩(15%)和间质纤维化;电镜下肾小管萎缩和间质纤维化,足细胞未见异常;病理诊断为微小病变。予足量泼尼松口服治疗4周无效,联合环磷酰胺冲击1疗程(累计150 mg·kg-1)后尿蛋白仍55~83 mg·kg-1。4年前出现多饮多尿,外院检查,血肌酐29.4 μmol·L-1,矫正肌酐清除率125 mL·(min·1.73 m2)-1;尿α1微球蛋白161 mg·L-1,尿微量白蛋白64 mg·L-1;尿蛋白电泳小分子蛋白占61.3%;4年期间低磷血症(0.62~0.89 mmol·L-1)、低钾血症(2.9~3.2 mmol·L-1)、低氯血症(91.8~95.4 mmol·L-1)、低镁血症(0.75~0.84 mmol·L-1),代谢性酸中毒(HCO3-15.6~20.7 mmol·L-1),泛氨基酸尿,未发现高钙尿症(24 h 0.05~0.08 mmol·kg-1);肾脏超声未见异常。临床诊断范可尼综合征、佝偻病。口服枸橼酸钾(2 mmol·kg-1·d-1)和碳酸氢钠(3 mmol·kg-1·d-1)治疗。近2年来间断手足搐搦。为进一步诊治来我院。
查体:身高110 cm,体重21 kg,均低于第3百分位,肋骨外翻,X形腿,余无特殊。
辅助检查:血肌酐54.2 μmol·L-1,矫正肌酐清除率102 mL·(min·1.73 m2)-1,血白蛋白46 mg·L-1,总胆固醇4.12 mmol·L-1;尿蛋白定量44~94 mg·kg-1;尿α1微球蛋白161~186 mg·L-1,尿微量白蛋白70~124 mg·L-1;尿蛋白电泳低分子蛋白占63.7%;血钾1.76 mmol·L-1,血镁0.76 mmol·L-1,血磷0.57 mmol·L-1,血氯91.3 mmol·L-1,血HCO3-23.6 mmol·L-1;尿比重1.005~1.008;24 h尿钙0.06 mmol·kg-1;泛氨基酸尿症。肾脏超声示肾钙化症。临床特征概括见表1,诊断为范可尼综合征、佝偻病。
例2 患儿男(例1胞弟),2.3岁,因“发现蛋白尿8个月”于2015年8月17日入住我院儿科肾脏病房。
患儿系G2P2,母孕期无特殊记载,出生史未见异常。8个月前尿检发现尿蛋白2+~3+,无伴随症状和自觉不适,未予处理。
查体:身高88 cm,体重12 kg,均低于第10百分位,余未见异常。
辅助检查:血肌酐25.8 μmol·L-1,矫正肌酐清除率116 mL·min-1·1.73 m-2,血白蛋白50 mg·L-1,总胆固醇3.26 mmol·L-1;尿蛋白定量85~98 mg·kg-1;尿α1微球蛋白413~708 mg·L-1,尿微量白蛋白318~470 mg·L-1;尿蛋白电泳低分子蛋白占56.0%;24 h尿钙0.23~0.25mmol·kg-1;泛氨基酸尿症;血电解质未见异常。肾脏超声未见异常。临床特征概括见表1,诊断为Dent病。
基因检测:经我院伦理委员会审批、患儿家长知情同意后,采用二代测序技术对2例患儿同时行遗传性肾小管疾病相关基因包突变分析,图1显示,2例患儿CLCN5基因第7外显子c.731C>T(p.S244L)半合子突变;母亲为携带者,临床无蛋白尿、血尿、高钙尿症;因其为X连锁遗传,父亲未行基因检测。遗传性范可尼综合征的其他相关基因如OCRL1、COQ2、CNTS、GALT1、ALDOB、SLC2A2、SLC5A2、ATP7B和NDUFAF6均未见异常。

表1 2例患儿临床特征总结
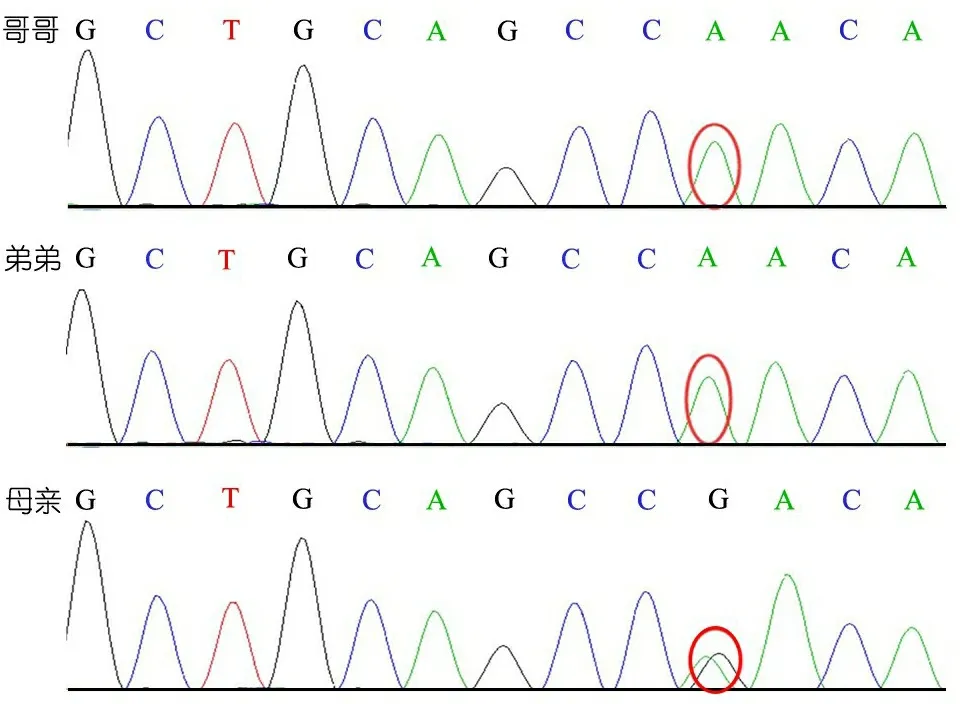
图1家系基因突变分析结果
注 2例患儿CLCN5基因第7外显子c.731C>T(p.S244L)半合子突变,母亲为杂合携带者
诊断明确后,例1给予枸橼酸钾(2~3 mmol·kg-1·d-1)、磷酸盐合剂(磷酸氢二钠和磷酸二氢钠,3~5 mmol·kg-1·d-1)口服治疗,随访1.5年,患儿血电解质紊乱恢复并维持正常,多饮多尿、蛋白尿、低比重尿均无明显变化,肾功能正常;例2给予氢氯噻嗪(1 mg·kg-1·d-1)和枸橼酸钾(2~3 mmol·kg-1·d-1)治疗,随访1.5年,患儿尿钙降至正常并维持(0.08~0.09 mmol·kg-1·d-1),尿蛋白无明显变化,肾功能正常。
2 讨论
Dent 病是一种罕见的X 连锁隐性遗传性的近端肾小管疾病,因CLCN5基因(Dent病Ⅰ型)和OCRL基因(Dent病Ⅱ型)突变所致[1, 2],临床主要特征为低分子蛋白尿、高钙尿症和肾脏钙化,随病程进展部分患者可出现肾功能异常或肾衰竭[3~5];还可以有其他多种表现,包括泛氨基酸尿、磷酸盐尿、糖尿症、高尿酸尿症、高钾尿症、血尿、尿酸化功能障碍、佝偻病或骨软化症等[6, 7]。
2/3的Dent病是由CLCN5基因突变所致。CLCN5基因编码的电压门控性 Cl-/H+离子交换蛋白(CLC-5)主要参与近曲小管胞吞途径介导的低分子蛋白的重吸收作用[3, 8]。目前约有260种CLCN5基因突变被报道,其中33%为错义突变、29%为移码突变、18%为无义突变、12%为剪切突变,8%其他突变[9]。
Dent病的临床表型在同一家系及不同家系之间变异均较大,CLCN5基因型和临床表型之间也无相关性[9,10]。目前已知,CLCN5基因突变是遗传性范可尼综合征的病因之一[11~15]。范可尼综合征是一组因近端肾小管的功能异常引起的症候群,尿中氨基酸、糖、磷酸盐、尿酸和碳酸氢盐等多种物质排泄过多。患者儿童期表现为生长发育迟缓、多饮、多尿、脱水和佝偻病,成人期表现为骨质疏松症和骨软化症,同时伴有多种电解质紊乱,包括低钾血症、代谢性酸中毒等,还有高钙尿症、低分子蛋白尿等。根据病因,范可尼综合征可分为遗传性、获得性和外源性[16, 17]。Dent病是遗传性肾性范可尼综合征的病因之一[13]。
本文报告的同一家系兄弟2例患儿,均有肾病水平的蛋白尿、泛氨基酸尿症,例1同时伴有多饮、多尿、低钾血症、低磷血症、代谢性酸中毒、低比重尿、肾脏钙化和佝偻病,但无高钙尿症,临床符合范可尼综合征;例2同时伴有高钙尿症,而无其他相关表现,临床符合Dent病。基因检测结果显示2例均有CLCN5基因半合子突变(c.731C>T ,p.S244L),均来自母亲(携带者)。该突变已有文献报道,为Dent病致病突变[18, 19]。为什么相同的CLCN5 基因突变导致不同的临床表型?
疾病的病程可能影响临床表型。例1(哥哥)5岁余发现蛋白尿,6岁余出现多饮、多尿,例2(弟弟)发现蛋白尿时仅1岁半。作为一种遗传性疾病,Dent病患儿生后不久即可出现低分子蛋白尿,而蛋白尿本身是肾脏预后不良的一个独立危险因素。哥哥病程相对较长,长期大量蛋白尿可能加重肾小管的损伤,从而临床表现多样,符合范可尼综合征。国内简珊等[14]报告的4例Dent病患儿(2例为CLCN5缺失突变,1例为CLCN5无义突变,1例未发现CLCN5和OCRL突变)中,病程超过5年的3例患儿临床均表现为范可尼综合征。提示随着病程进展,Dent病的小管病变逐渐加重,多饮、多尿、低比重尿和肾功能不全等的发生率逐渐增加[15]。当然,病程并不是唯一影响因素,如Hodgin[11]等报告的Dent病两兄弟(未行CLCN5和OCRL基因突变检测),年龄分别为6岁和4岁,起病时均表现为范可尼综合征,提示除了病程,可能还有其他因素影响Dent病的临床表型。
基因型对临床表型的影响。目前已知,无论在同一家系或不同家系,Dent病的基因型和临床表型之间似乎均无相关性[20]。本文患儿的CLCN5基因突变(c.731C>T,p.S244L)已经有文献报道,所有患儿临床都表现为典型Dent病,而未见表现为范可尼综合征者[18, 19]。这种同一家系中基因突变类型相同而临床表型不同的现象,也曾有文献报道,如Mansour-Hendili等[9]报告,同一家系中的Dent病患者,有些表现为范可尼综合征,伴或不伴佝偻病;另一些表现为低分子蛋白尿和高钙尿症,伴或不伴肾脏钙化或肾结石。此外,本文2例患儿均进行了遗传性范可尼综合征的其他相关致病基因检测,均未见异常,除外了合并遗传因素影响临床表型的可能。
肾脏病理对临床表型的影响。本文例1患儿5岁时肾脏病理结果显示肾小球微小病变伴明显的肾小管、间质病变,与临床表型较为一致。而且未发现有可能损伤肾小管、间质类的药物应用史。遗憾的是,例2患儿未行肾脏病理检查,不清楚其肾小管、间质病变的程度和范围。已有研究显示,对于Dent病患者,球性肾小球硬化、足突融合和间质炎性病变的比例越高,估算肾小球滤过率越低,而且足突融合与估算肾小球滤过率的逐年下降相关[21]。但是,目前尚没有Dent病肾小管、间质病变程度和临床表型相关性的研究。
CLCN5 基因突变导致不同临床表现的确切机制尚不完全清楚。作为一种电压门控性 Cl-/H+离子交换蛋白,CLC-5蛋白通过促进Cl-内流以中和生电性H+-ATPase泵出的H+,消除阳性跨膜电压,维持内吞体电中性,使H+不断泵入,从而使得内吞体持续维持酸性环境,通过胞吞途径对低分子蛋白等进行重吸收。CLCN5基因突变的Dent病患儿中,CLC-5蛋白异常,近曲小管重吸收功能障碍,出现低分子蛋白尿、泛氨基酸尿、糖尿症和磷酸盐尿症等。但是,高钙尿症和肾脏钙化的机制尚不明确。已知30%的CLCN5基因突变的患者临床无高钙尿症,但部分人却有肾脏钙化[22],这和本文例1相似。推测CLCN5基因突变时CLC-5蛋白功能缺失表现为近曲小管重吸收功能障碍,而肾脏结石形成通过其他可能的不同途径,提示CLCN5基因可能涉及小管的其他特殊功能[23]。这些机制的明确有助于解释不同临床表型的差异。
[1]Utsch B, Bokenkamp A, Benz MR, et al. Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis, 2006, 48(6): 942 e1-14
[2]Grand T, L'Hoste S, Mordasini D, et al. Heterogeneity in the processing of CLCN5 mutants related to Dent disease. Hum Mutat, 2011, 32(4): 476-483
[3]Devuyst O. Dent's disease: chloride-proton exchange controls proximal tubule endocytosis. Nephrol Dial Transplant, 2010, 25(12): 3832-3835
[4]Cho HY, Lee BH, Choi HJ, et al. Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol, 2008, 23(2): 243-249
[5]Annigeri RA, Rajagopalan R. Hypophosphatemic rickets due to Dent's disease: A case report and review of literature. Indian J Nephrol, 2009, 19(4): 163-166
[6]Wrong OM, Norden AG, Feest TG. Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM, 1994, 87(8): 473-493
[7]Frymoyer PA, Scheinman SJ, Dunham PB, et al. X-linked recessive nephrolithiasis with renal failure. N Engl J Med, 1991, 325(10): 681-686
[8]Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V. Dent's disease: clinical features and molecular basis. Pediatr Nephrol, 26(5): 693-704
[9]Mansour-Hendili L, Blanchard A, Le Pottier N, et al. Mutation Update of the CLCN5 Gene Responsible for Dent Disease 1. Hum Mutat, 2015, 36(8): 743-752
[10]Wong W, Poke G, Stack M, et al. Phenotypic variability of Dent disease in a large New Zealand kindred. Pediatr Nephrol, 2017, 32(2): 365-369
[11]Hodgin JB, Corey HE, Kaplan BS, et al. Dent disease presenting as partial Fanconi syndrome and hypercalciuria. Kidney Int, 2008, 73(11): 1320-1323
[12]Pook MA, Wrong O, Wooding C, et al. Dent's disease, a renal Fanconi syndrome with nephrocalcinosis and kidney stones, is associated with a microdeletion involving DXS255 and maps to Xp11. 22. Hum Mol Genet, 1993, 2(12): 2129-2134
[13]Solano A, Lew SQ, Ing TS. Dent-Wrong disease and other rare causes of the Fanconi syndrome. Clin Kidney J, 7(4): 344-347
[14]简珊, 魏珉, 何艳燕, 等. Dent 病 4 例临床及基因分析. 中国当代儿科杂志, 2015, 17(12): 1261-1266
[15]Bhardwaj S, Thergaonkar R, Sinha A, et al. Phenotype of Dent Disease in a Cohort of Indian Children. Indian Pediatr, 2016, 53(11): 977-982
[16]Maher OM, Moonat HR. Fanconi Anemia and Fanconi Syndrome: Time to Correct the Misnomers. J Pediatr Hematol Oncol, 2016, 38(7): 585
[17]Klootwijk ED, Reichold M, Unwin RJ, et al. Renal Fanconi syndrome: taking a proximal look at the nephron. Nephrol Dial Transplant, 2015, 30(9): 1456-1460
[18]Lloyd SE, Pearce SH, Fisher SE, et al. A common molecular basis for three inherited kidney stone diseases. Nature, 1996, 379(6564): 445-449
[19]Wu F, Reed AA, Williams SE, et al. Mutational analysis of CLC-5, cofilin and CLC-4 in patients with Dent's disease. Nephron Physiol, 2009, 112(4): p53-62
[20]Ludwig M, Utsch B, Monnens LA. Recent advances in understanding the clinical and genetic heterogeneity of Dent's disease. Nephrol Dial Transplant, 2006, 21(10): 2708-2717
[21]Wang X, Anglani F, Beara-Lasic L, et al. Glomerular Pathology in Dent Disease and Its Association with Kidney Function. Clin J Am Soc Nephrol, 2016, 11(12): 2168-2176
[22]Ludwig M, Utsch B, Balluch B, et al. Hypercalciuria in patients with CLCN5 mutations. Pediatr Nephrol, 2006, 21(9): 1241-1250
[23]Devuyst O, Luciani A. Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J Physiol, 2015, 593(18): 4151-4164
