Photocatalytic Production of Hydrogen Peroxide Using g-C3N4 Coated MgO-Al2O3-Fe2O3 Heterojunction Catalysts Prepared by a Novel Molten Salt-Assisted Microwave Process
2018-01-15CHENXinHUShaoZhengLIPingLIWeiMAHongFeiLUGuang
CHEN Xin HU Shao-Zheng* LI Ping LI Wei MA Hong-Fei LU Guang
Photocatalytic Production of Hydrogen Peroxide Using g-C3N4Coated MgO-Al2O3-Fe2O3Heterojunction Catalysts Prepared by a Novel Molten Salt-Assisted Microwave Process
CHEN Xin HU Shao-Zheng* LI Ping LI Wei MA Hong-Fei LU Guang
()
H2O2is industrially produced by the anthraquinone method, in which energy consumption is high because it involves multistep hydrogenation and oxidation reactions. Photocatalytic production of H2O2has received increasing attention as a sustainable and eco-friendly alternative to conventional anthraquinone-based and electrochemical production processes. Herein, we report a novel molten salt-assisted microwave process for the synthesis of a g-C3N4-coated MgO-Al2O3-Fe2O3(MAFO) heterojunction photocatalyst with outstanding H2O2production ability. The addition of a molten salt during synthesis changes the morphology of the as-prepared catalysts and influences the degree of polycondensation of melamine, leading to a change in the band gap energy. The cladding structure forms the maximum area of the heterojunction, leading to strong electronic coupling between the two components.This strong electronic coupling results in amoreeffective separation of the photogeneratedelectron-hole pairs and a faster interfacial charge transfer, leading to higher H2O2formation rate. The equilibrium concentration and formation rate of H2O2over the as-prepared heterojunction catalyst were 6.3 mmol·L−1and 1.42 mmol·L−1·h−1, which are much higher than that reported for g-C3N4and MAFO individually. In addition, the H2O2decomposition rate also decreases over the as-prepared heterojunction catalysts. A possible mechanism and the electron transfer routes have been proposed based on a free radical trapping experiment.
g-C3N4; Cladding structure; Heterojunction; H2O2production; Molten salt-assisted microwave process
1 Introduction
Hydrogen peroxide (H2O2), as a highly efficient and green oxidant, has been widely used in bleaching and disinfectant applications, such as textile, paper pulp and medical industry. H2O2has the highest content of active oxygen (47%,/) and only H2O as the by-product1,2. Besides that, H2O2is also an ideal energy carrier alternative to hydrogen with the low volumetric energy3,4. The output potential of H2O2fuel cell is 1.09 V theoretically, which is comparable with that of hydrogen fuel cell (1.23 V)5–7. In the industry, H2O2is produced by the anthraquinone method, in which energy consumption is high because of the multistep hydrogenation and oxidation reactions. Recently, direct synthesis of H2O2from only water, oxygen and visible light through two-electron reduction from the conduction band has been widely studied using semiconductor as photocatalyst (Reaction (1))8–10. Holes in the valence band (VB) can directly oxidize water molecules to produce O2(Reaction (2)). But usually, hole scavenger such as alcohols are added to promote the electrons-holes separation rate (Reaction (3)). This method has many advantages such as green cleaning, mild conditions and low power consumption. However, the H2O2can be decomposed by the reduction with e-which causes the H2O2equilibrium concentration is not satisfactory till now (Reaction (4)).

O2 + 2H+ + 2e−→ H2O2(1) H2O + 2h+→ 1/2 O2 + 2H+(2) R-OH + 2h+→ Oxidation product(3) H2O2 + e−→ •OH + OH−(4)
Recently, graphitic carbon nitride (g-C3N4), a metal-free visible light photocatalyst, has received increasing attention due to its suitable electric band-gap, good chemical stability, and unique electronic structure. Besides that, its conduction band potential is ~ −1.3 V, more negative than the reduction potential of O2/H2O2(0.695 V). Thus g-C3N4can reduce O2to H2O2thermodynamically10. However, the low separation efficiency of photogenerated electron-hole pairs and the poor visible light utilization limits its practical applications. In order to improve the above situation, researchers have developed many strategies, including tailoring microstructures11,12, forming surface defects13,14, doping15,16and building heterojunctions17,18. These strategies mainly focus on shortening the distance of transfer paths, forming unique transmission channels, offering more active sites for trapping carriers, and facilitating the separation and transmission of photogenerated electron-hole pairs. Metal oxides and sulphides are widely used to build the heterojunction with g-C3N4. In addition to single metal sulfide, some multi-metal sulfides coupled g-C3N4composites are also reported19–21. However, few studies concerning multi-metal oxide (MMO) coupled g-C3N4composites are reported22,23. With the tunable composition, MMO possesses the special optical properties and electronic structure, leading to the formation of tunable band structure. This is beneficial to the energy level matching of two semiconductors, which is significant important to form the heterojunction.
Molten salt method is widely used in the materials synthesis field in recent years because it can accelerate diffusion of constituent ions, control crystal growth and easily separate from the solid product by dissolving in water24–26. In general, molten salt can serve as a reaction medium for reactant dissolution and precipitation, as soft template for tailoring micro and mesoporosity of the materials, and as structure- directing agent in the polycondensation and deamination reaction to obtain graphitic materials. The features of this synthesis method are related to the surface and interface energies between the constituents and the salt, resulting in a tendency to minimize the energies by forming a specific morphology. Though the electric-resistance heating molten-salt method can be used to synthesize g-C3N4photocatalysts, the problems of long time consuming and high energy consumption in synthesis, large emission of harmful gas and low catalyst yield are still difficult to overcome27.
Recently, microwave-assisted heating synthesis has also been widely used to prepare nanomaterials28–30. The microwave treatment can transfer energy from microwave to the microwave-absorber material which induces strong heating in minutes. When the microwave energy is absorbed by the raw material, the molecules are orderly arrangement in the electromagnetic field of the microwave. Then the high frequency reciprocating motion occurs inside the molecules of raw materials, causes the frequent collisions between molecules, leading to the generation of a lot of frictional heat. Under this heating method, the raw material is rapidly heated without the presence of temperature gradient. It is reported that g-C3N4can be synthesized by the microwave-assisted heating method in a few minutes, suggesting this is a potential way in rapid synthesis of carbon nitride based materials31. Besides that, the catalyst yield is high and the emission of harmful gas is also low with this method. It is known that the solid-state reaction system needs a uniform reaction medium to achieve a stable and reliable condition. The molten-salt process can offer a unique liquid condition that is stable and convenient for the solid-state reaction system. Thus, we hypothesize that combination two methods mentioned above should be an effective strategy to synthesize g-C3N4based materials with high performance. In this work, the g-C3N4coated MgO-Al2O3- Fe2O3(MgAlFeO) heterojunction catalysts were synthesized via a novel molten salt-assisted microwave process. The photocatalytic activities were evaluated in the photocatalytic H2O2production under visible light. The possible mechanism is proposed.
2 Experimental
2.1 Preparation and characterization
All the chemicals used in this experiment were reagent gradeand without further treatment. Polymetallic oxide MgAlFeO was prepared as follow. Mixed salt solutions of Mg(NO3)2·6H2O, Al(NO3)3·6H2O and Fe(NO3)3·9H2O (molar ratio of Mg : Al : Fe is 5 : 2 : 1) were added into 80 mL deionized water under stirring. Then, desired amount of urea (molar ratio urea/total metal = 2) was added. The obtained solution was placed in a stainless autoclave, which has a 100 ml Teflon inner liner. The autoclave was sealed, placed in an oven and maintained at 120 °C for 8 h. The solid was collected by centrifugation, washed with deionized water fore three times, dried at 70 °C overnight and denoted as MAFO.
In a typical process, eutectic mixture of KCl-LiCl (1 : 1 weight ratio) was selected as a solvent with the melting point of 350 °C. The mixture of eutectic salts, melamine and MAFO (with a weight ratio of 50 : 2 : 1) were finely ground in a mortar, then transferred to an alumina crucible and treated by microwave for 20 min in a normal microwave oven (G70D20CN1P-D2, Galanz). The input power of microwave oven is 1.0 kW·h−1. The obtained catalyst was denoted as MV-MS-CN/MAFO, in which MVand MSstand for microwave treatment and molten salt assistant respectively. For comparison, MV-CN, MV-MS-CN and MV-CN/MAFO were prepared following the same procedure as in the synthesis of MV-MS-CN/MAFO but in the absence of eutectic salts and MAFO, MAFO, and eutectic salts, respectively. Bulk g-C3N4was prepared by heating melamine at 550 °C for 4 h at the rate of 5 °C·min−1. The product was denoted as B-CN.
The XRD patterns of the prepared samples were recorded on a Rigaku D/max-2400 instrument (Shimadzu, Japan) using Cu-Kradiation (= 0.154 nm). The scan rate, step size, voltage and current were 0.05 (°)·min−1, 0.01°, 40 kV and 30 mA, respectively. UV-Vis spectroscopy was carried out on a V-550 model UV-Vis spectrophotometer (JASCO Japan) using BaSO4as the reflectance sample. The morphologies of prepared catalyst were observed by using a scanning electron microscope (SEM, JSM 5600LV, JEOL Ltd. Japan). TEM images were taken on a Philips Tecnai G220 model microscope (Holand). Nitrogen adsorption was measured at −196 °C on a Micromeritics 2010 analyser (USA). All the samples were degassed at 393 K prior to the measurement. The BET surface area (BET) was calculated based on the adsorption isotherm. The pore-size distributions were obtained from the desorption branches using the Barrett-Joyner-Halenda (BJH) method. ICP was performed on a Perkin-Elmer Optima 3300DV apparatus (USA). The photoluminescence (PL) spectra were measured at room temperature with a fluorospectrophotometer (JASCO FP-6300, Japan) using a Xe lamp as the excitation source. The electrochemical impedance spectra (EIS) were recorded using an EIS spectrometer (EC-Lab SP-150, BioLogic Science Instruments, USA) in a three electrode cell by applying a 10 mV alternative signal versus the reference electrode (SCE) over a frequency range of 1 MHz to 100 mHz. The cyclic voltammograms were measured in a 0.1 mol∙L−1KCl solution containing 2.5 mmol∙L−1K3[Fe(CN)6]/K4[Fe(CN)6] (1 : 1) as a redox probe at a scanning rate of 20 mV·s−1in the same three electrode cell as the EIS measurement.
2.2 Photocatalytic reaction
The photocatalytic H2O2production ability of the samples were evaluated by the reduction of molecular oxygen. For these experiments, 0.2 g of photocatalyst was added to 200 mL deionized water. The suspension was dispersed using an ultrasonicator for 10 min. During the photoreaction under visible light irradiation, the suspension was exposed to a 250 W high-pressure sodium lamp with main emission in the range of 400 to 800 nm, and O2was bubbled at 80 mL∙min−1through the solution. The UV light portion of the sodium lamp was filtered by a 0.5 mol∙L−1NaNO2solution. All runs were conducted at ambient pressure and 30 °C. At given time intervals, 5 mL aliquots of the suspension were collected and immediately centrifuged to separate the liquid samples from the solid catalyst. The concentration of H2O2was analyzed by normal iodometric method32,33.

Fig.1 XRD patterns (a), N2 adsorption-desorption isotherms (b), UV-Vis spectra (c) and plots of the transformed Kubelka-Munk function versus the energy of light (d) of as-prepared catalyst.
3 Results and discussion
Fig.1(a) shows the XRD patterns of as-prepared catalysts. Two typical diffraction peaks of g-C3N4are present in the B-CN, MV-CN and MV-MS-CN. The peak at 13.1°corresponds to in-plane structural packing motif of tri-s-triazine units, which is indexed as (100) peak. The peak at 27.5° corresponds to interlayer stacking of aromatic segments with distance of 0.324 nm, which is indexed as (002) peak. For MAFO, six diffraction peaks are observed, which are assigned to MgO, Al2O3and Fe2O3, respectively34–36. In the case of as-prepared heterojunction catalysts, both diffraction peaks for g-C3N4and MAFO are observed. Neither diffraction peak of other species nor the diffraction peak shift is observed indicating that no doping occurs. To characterize the specific surface area of as-prepared catalysts, the nitrogen adsorption and desorption isotherms were measured (Fig.1(b)). The isotherms of all the catalysts are of classical type IV, suggesting the presence of mesopores. The BET specific surface areas (BET) of B-CN, MV-CN, MV-MS-CN, MAFO, MV-CN/MAFO and MV-MS-CN/ MAFO are calculated to be 6.5, 7.2, 14.9, 10.6, 8.8 and 13.7 m2·g−1. This hints that the morphology of MV-MS-CN could be changed by addition of molten salt in the synthesis process, leading to the significantly promotedBET. The largeBETcan promote adsorption, desorption and diffusion of reactants and products, which is favorable to the photocatalytic performance.

Table 1 The components of as-prepared catalysts obtained by ICP.
The UV-Vis spectra of the as-prepared photocatalysts are shown in Fig.1(c). The band gaps are estimated from the tangent lines in the plots of the square root of the Kubelka-Munk function as a function of the photon energy (Fig.1(d))37. B-CN displays an absorption edge at approximately 450 nm, corresponding to a band gap of 2.75 eV. The absorption edge for MV-CN and MV-MS-CN are 480 and 491 nm, and the corresponding band gap is estimated to be 2.58 and 2.52 eV. This reveals that both microwave and molten salt can influence the polycondensation degree of melamine, leading to the change of band gap energy. MAFO shows obviously improved visible light absorption ability compared with as-prepared g-C3N4catalysts. Its absorption edge is observed at 647 nm, and the corresponding band gap is estimated to be 1.92 eV. For the as-prepared heterojunction photocatalysts, the typical two absorption edges for g-C3N4and MgAlFeO are observed, hinting the heterojunction photocatalysts are composed of these two components. No intrinsical difference between MV-CN/MAFO and MV-MS-CN/ MAFO can be observed.
The components of as-prepared catalysts were obtained by ICP (Table 1). The C and N contents for as-prepared g-C3N4catalysts are approximately 39% and 57% (, mass fraction), which is close to the theoretical values. For MAFO, the Mg, Al, Fe and O contents are 31.5%, 14.1%, 14.7% and 39.7% (), respectively. Both the as-prepared heterojunction photocatalysts show the similar element percentages, indicating that the addition of molten salt do not influence the component of catalyst. According to this element percentage, the mass ratio of g-C3N4to MgAlFeO is approximately 6 : 4 for the as-prepared heterojunction photocatalysts. In addition, no K, Li and Cl are detected in the as-prepared catalysts, confirming that no ion-doping occurs.

Fig.2 SEM images of as-prepared B-CN (a), MAFO (b), MV-CN (c), MV-MS-CN (d), MV-CN/MAFO (e), MV-MS-CN/MAFO (f and g) and HRTEM of MV-MS-CN/MAFO (h).
The morphologies of the representative samples were examined by using SEM analysis. Fig.2a indicates that as-prepared B-CN is composed of a large number of irregular particles. Those particles exhibit layer structure that is similar to its analogue graphite. In Fig.2b, the nanorod-like MAFO with ~2 µm long is observed. Besides that, it is noted that the surface of MAFO is smooth. For MV-CN, the catalyst with layered structure is still observed (Fig.2c). In the case of MV-MS-CN, the catalyst morphology changes to nanoparticles (Fig.2d). This confirms that the addition of molten salt significantly influence the morphology of as-prepared catalyst. Those nanoparticles could form more intra-aggregated pores, leading to the increasedBET(Fig.1b). Fig.2e clearly displays that the as-prepared MV-CN/MAFO is composed of layer structural MV-CN and nanorod-like MAFO. MAFO nanorods seem to stick to the MV-CN surface. This interaction between MV-CN and MAFO is poor. For MV-MS-CN/MAFO (Fig.2f), it can be seen that the nanorod-like MAFO is coated by the MV-MS-CN nanoparticles. The MAFO surface is not as smooth as shown in Fig.2b but very rough, confirming the MV-MS-CN coating (Fig.2g). High-resolution transmission electron microscopy (HRTEM) analysis was performed to get information on the microstructure of as-prepared MV-MS-CN/ MAFO (Fig.2h). The observed lattice fringe spacing of 0.324 nm corresponds to the (002) crystal plane of g-C3N4(JCPDS 87-1526). The lattice fringes of 0.373, 0.265 and 0.208 nm in the HRTEM image should be assigned to the (012), (006) and (200) planes of Al2O3, Fe2O3and MgO, respectively. Smooth and intimate interfaces are clearly observed between MV-MS-CN and MAFO, which confirms the formation of g-C3N4/MgAlFeO heterojunction. Such strong interaction can result in the higher interfacial charge transfer rate and H2O2production ability.
XP spectra are used to investigate the structure of the as-prepared heterojunction catalyst. In Mg 2, Al 2and Fe 2regions (Fig.3(a–c)), the binding energies for MAFO located at 49.7, 73.8 and 711.4 eV are assigned to the Mg2+, Al3+and Fe3+respectively38–40. For MV-MS-CN/MAFO, the binding energies in Mg 2, Al 2and Fe 2regions exhibit obvious blue-shifts compared with that of MAFO. This is probably due to the electron transfer from electron-rich g-C3N4to MAFO, leading to the electron density change. In O 1region (Fig.3d), the MAFO displays a single peak at 531.5 eV, which is assigned to the O2−bond to the metal ions. For MV-MS-CN/MAFO, another peak located at 532.6 eV is observed. As reported by previous literatures, this peak should be assigned to the adsorbed oxygen species41–43.In Fig.3e, the spectra of both two catalystsin C 1regions can be fitted with two contributions which located at 284.6, and 287.8 eV. The sharp peak around 284.6 eV is attributed to the pure graphitic species in the CN matrix. The peak with binding energy of 287.8 eV indicates the presence of2C atoms bonded to aliphatic amine (―NH2or ―NH―) in the aromatic rings44.In Fig.3(f), the main N 1peak of MV-MS- CN located at 398.3 eV can be assigned to2-hybridized nitrogen (C=N―C), thus confirming the presence of2-bonded graphitic carbon nitride. The peak at a higher binding energy of 400.1 eV is attributed to tertiary nitrogen (N―(C)3) groups45. For MV-MS-CN/MAFO, the 0.2 eV shift to higher binding energy is observed, indicating the decreased electron density of nitrogen atoms. Combined with the phenomenon of binding energy shift in Mg 2, Al 2and Fe 2regions, it is deduced that the strong electronic interaction between the MV-MS-CN and MAFO is formed in MV-MS-CN/ MAFO.
The energy level positions of MV-MS-CN and MAFO are confirmed by VB XPS. In Fig.4a, the VB positions of MV-MS-CN and MAFO are +1.29 and +2.08 V. It is obtained from the UV-Vis results that the band gaps for CN and MgAlFeO are 2.52 and 1.92 eV. Thus theCBfor MV-MS-CN and MAFO is −1.23 and +0.16 V, respectively. Once MV-MS- CN and MAFO are electronically coupled together, the band alignment between the two components results in the formation of heterojunction with well-matched band structure. The potential difference is the main driving force for efficient charge separation and transfer. These two charge transfer processes are beneficial for overcoming the high dissociation barrier of the Frenkel exciton and stabilizing electrons and holes. As the photogenerated electrons and holes are spatially separated into two components, the charge recombination is drastically inhibited, which is of great benefit for enhancing the photocatalytic activity. In addition, with effective separation of electron/hole pairs, the lifetime of photogenerated charge carriers is expected to be prolonged. The prolonged lifetime allows the fast charge transfer to the reactive substrates on the photocatalyst surface, promoting the photocatalysis reaction.
EIS was used to characterize charge-carrier migration and confirm the interfacial charge transfer effect of the as-prepared heterojunction catalysts. As shown in Fig.4b, the as-prepared heterojunction catalysts exhibit a decreased arc radius compared to that of MV-MS-CN and MAFO. In general, the radius of the arc in the EIS spectra reflects the reaction rate on the surface of the electrode46.The reduced arc radius indicates a diminished resistance of the working electrodes, suggesting a decrease in the solid-state interface layer resistance and the charge transfer resistance across the solid-liquid junction on the surface between g-C3N4and MgAlFeO47,48.MV-MS-CN/ MAFO displays the smallest arc radius, confirming that a more effective separation of photogenerated electron-hole pairs and a faster interfacial charge transfer occur on the MV-MS-CN/ MAFO surface compared with MV-CN/MAFO. PL spectra are shown in Fig.4c. In general, the higher PL intensity, the lower separation rate of electrons-holes. Obviously, the PL intensity follows the order: MAFO > MV-MS-CN > MV-CN/MAFO > MV-MS-CN/MAFO, which is consistent with the order of arc radius of EIS spectra.MV-MS-CN/MAFO exhibits a significant fluorescence quenching phenomenon, indicating its significantly improved separation efficiency. This confirms the existence of strong interaction between MV-MS-CN and MAFO.

Fig.4 VB XPS (a), EIS (b) and PL (c) of as-prepared catalysts.

Table 2 Influences of molar ratio of Mg : Al : Fe and mass ratio of melamine to MAFO on the H2O2 production performance.
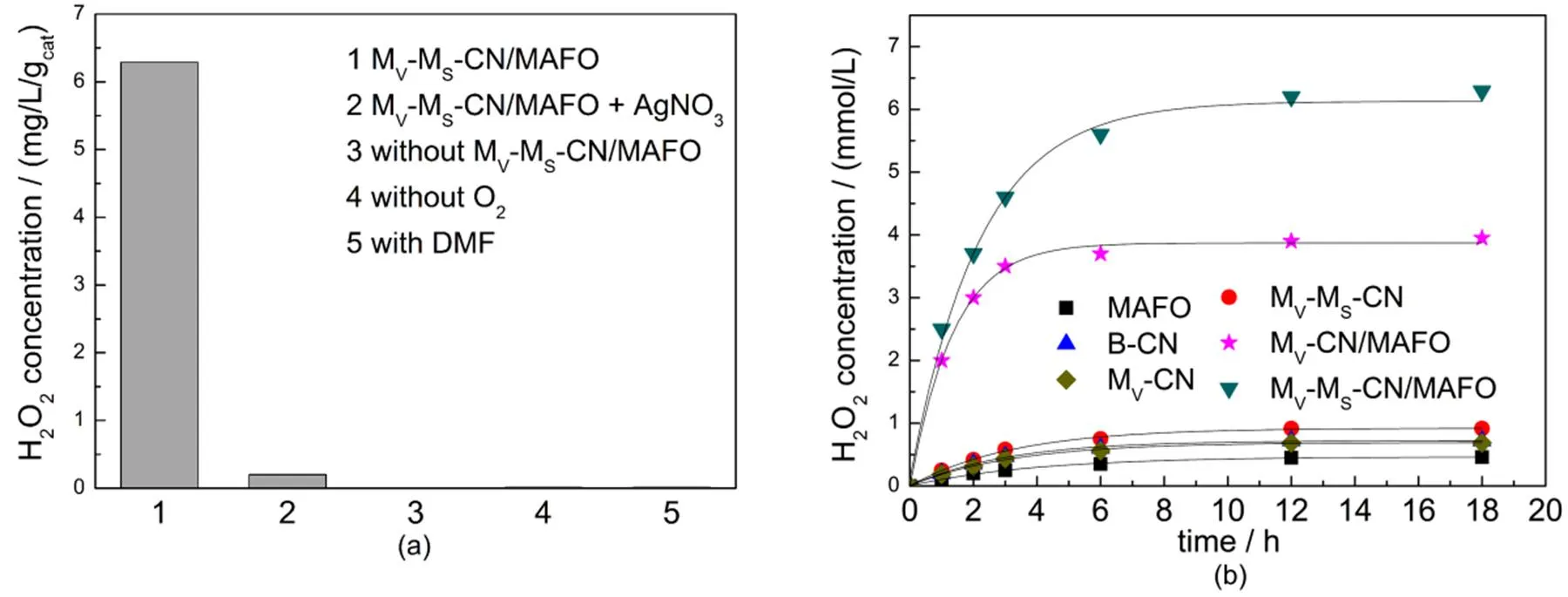
Fig.5 H2O2 production ability of MV-MS-CN/MAFO under different reaction conditions (a) and the H2O2 production ability over as-prepared catalysts (b).
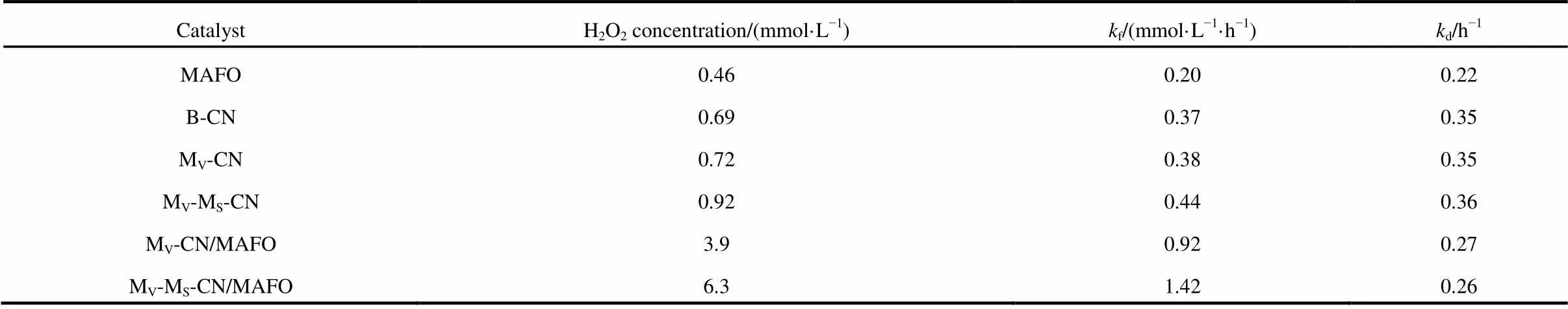
Table 3 H2O2 concentration and the kinetic parameters of as-prepared catalysts.
The catalytic stability of MV-MS-CN/MAFO is shown in Fig.6. It is clearly seen that the H2O2concentration reaches a constant level at 6 h. After 48 h, the equilibrium concentration of H2O2is almost unchanged, hinting its excellent catalytic stability. Fig.6 inset shows the XRD results of fresh and reused MV-MS-CN/MAFO. No obvious difference between them is shown, confirming its outstanding structure stability.
The catalytic stability of MV-MS-CN/MAFO is shown in Fig.6. It is clearly seen that the H2O2concentration reaches a constant level at 6 h. After 48 h, the equilibrium concentration of H2O2is almost unchanged, hinting its excellent catalytic stability. Fig.6 inset shows the XRD results of fresh and reused MV-MS-CN/MAFO. No obvious difference between them is shown, confirming its outstanding structure stability.

Fig.6 Catalytic stability of MV-MS-CN/MAFO.

Fig.7 Photocatalytic RhB degradation performance (a) and the possible electrons transfer route (b) over MV-MS-CN/MAFO.
In order to clarify the type of heterojunction, photocatalytic RhB degradation performance over MV-MS-CN/MAFO is investigated (Fig.7(a)).-BuOH was used as hydroxyl radical (•OH) scavenger. The results indicate that the photocatalytic degradation rate of RhB is over 80% within 4 h. When t-BuOH is added, the degradation rate of RhB sharply decreases to approximately 50%. This hints hydroxyl radical is one of the main oxidative species for RhB degradation. It is known that the redox potentials for •OH/OH−is +1.99 V51. TheVBof MV-MS-CN and MAFO is +1.29 and +2.08 V, respectively. Thus, the holes in valence band of MAFO are positive enough to generate •OH but MV-MS-CN cannot. It is deduced that the holes are in the valence band of MAFO but not MV-MS-CN. Accordingly, the heterojunction should be “Z” type. The possible electron transfer mechanism is shown in Fig.7(b). Under visible light irradiation, the electrons-holes are formed. The electrons in the conduction band of MAFO transfer to the valence band of MV-MS-CN to combine with the holes. Thus, the electrons in the conduction band of MV-MS-CN will reduce oxygen to form H2O2: O2+ 2H++ 2e−→ H2O2. Holes in the VB of MAFO can oxidize water to form oxygen as follow:
H2O + 2h+→ 1/2 O2+ 2H+52.
4 Conclusions
In this work, the g-C3N4coated MgAlFeO heterojunction catalyst was synthesized via a novel molten salt-assisted microwave process. The addition of molten salt into synthesis process not only changes the morphology of as-prepared catalysts but also influence the polycondensation degree of melamine, leading to the change of band gap energy. SEM and HRTEM results show that the MgAlFeO nanorods are coated by the g-C3N4nanoparticles, leading to the strong electronic coupling between two components. This strong electronic coupling results inmore effective separation of photogenerated electron-hole pairs and faster interfacial charge transfer, causing the higher H2O2formation rate (f). In addition, the VB holes of MAFO are positive enough to generate •OH which is the H2O2decomposition product, leading to the lower H2O2decomposition rate (d) over as-prepared heterojunction catalysts. This work provides a novel method to prepare heterojunction catalyst with high electron-hole separation rate and photocatalytic performance.
(1) Campos-Martin, J. M.; Blanco-Brieva, G.; Fierro, J. L. G.. 2006,, 6962. doi: 10.1002/anie.200503779
(2) Samanta, C.2008,, 133. doi: 10.1016/j.apcata.2008.07.043
(3) Yamazaki, S.; Siroma, Z.; Senoh, H.; Ioroi T.; Fujiwara, N.; Yasuda, K.2008,, 20. doi: 10.1016/j.jpowsour.2007.12.013
(4) Shaegh, S. A. M.; Nguyen, N. T.; Ehteshami, S. M. M.; Chan, S. H.. 2012,, 8225. doi: 10.1039/C2EE21806B
(5) Yamada, Y.; Fukunishi, Y.; Yamazaki, S.; Fukuzumi, S.. 2010,, 7334. doi: 10.1039/c0cc01797c
(6) Yamada, Y.; Yoshida, S.; Honda, T.; Fukuzumi, S.. 2011,, 2822. doi: 10.1039/C1EE01587G
(7) Kato, S.; Jung, J.; Suenobua, T.; Fukuzumi, S.. 2013,, 3756. doi: 10.1039/C3EE42815J
(8) Tsukamoto, D.; Shiro, A.; Shiraishi, Y.; Sugano, Y.; Ichikawa, S.; Tanaka, S.; Hirai, T.. 2012,, 599. doi: 10.1021/cs2006873
(9) Diesen, V.; Jonsson, M.. 2014,, 10083. doi: 10.1021/jp500315u
(10) Li, S.; Dong, G. H.; Hailili, R.; Yang, L. P.; Li, Y. X.; Wang, F.; Zeng, Y. B.; Wang, C. Y.. 2016,, 26. doi: 10.1016/j.apcatb.2016.03.004
(11) Kong, H. J.; Won, D. H.; Kim, J.; Woo, S. I.. 2016,, 1318. doi: 10.1021/acs.chemmater.5b04178
(12) Wang, Z.; Guan, W.; Sun, Y.; Dong, F.; Zhou, Y.; Ho, W. K.2015,, 2471. doi: 10.1039/c4nr05732e
(13) Yang, P.; Zhao, J.; Qiao, W.; Li, L.; Zhu, Z.2015,, 18887. doi: 10.1039/c5nr05570a
(14) Kang, Y.; Yang, Y.; Yin, L. C.; Kang, X.; Liu, G.; Cheng, H. M.. 2015,, 4572. doi: 10.1002/adma.201501939
(15) Hu, S. Z.; Chen, X.; Li, Q.; Li, F. Y.; Fan, Z. P.; Wang, H.; Wang, Y. J. Zheng, B. H.; Wu, G.. 2017,, 58. doi: 10.1016/j.apcatb.2016.08.002
(16) Fan, X.; Zhang, L.; Wang, M.; Huang, W.; Zhou, Y.; Li, M.; Cheng, R.; Shi, J.. 2016,, 68. doi: 10.1016/j.apcatb.2015.09.006
(17) Zhang, Q.; Hu, S. Z.; Fan, Z. P.; Liu, D. S.; Zhao, Y. F.; Ma, H. F.; Li, F. Y.. 2016,, 3497. doi: 10.1039/c5dt04901f
(18) Zhu, Z.; Lu, Z.; Wang, D.; Tang, X.; Yan, Y.; Shi, W.; Wang, Y.; Gao, N.; Yao, X.; Dong, H.. 2016,, 115. doi: 10.1016/j.apcatb.2015.09.029
(19) Hu, S. Z.; Li, Y. M.; Li, F. Y.; Fan, Z. P.; Ma, H. F.; Li, W.; Kang, X. X., 2269. doi: 10.1021/acssuschemeng.5b01742
(20) Nie, Q.;Yuan, Q.; Wang, Q.. 2004,, 5611. doi: 10.1023/B:JMSC.0000039301.70811.a4
(21) Fu, X. L.; Wang, X. X.; Chen, Z. X.; Zhang, Z. Z.; Li, Z. H.; Leung, D. Y. C.; Wu, L.; Fu, X. Z.. 2010,, 393. doi: 10.1016/j.apcatb.2010.01.018
(22) Chen, J.; Shen, S. H.; Guo, P. H.; Wu, P.; Guo, L. J.2014,, 4605. doi: 10.1039/C3TA14811D
(23) Lan, M.; Fan, G. L.; Yang, L.; Li, F..2015,, 5725. doi: 10.1039/C4RA07073A
(24) Bojdys, M. J.; Muller, J.; Antonietti, M.; Thomas, A.. 2008,, 8177. doi: 10.1002/chem.200800190
(25) Wirnhier, E.; Doblinger, M.; Gunzelmann, D.; Senker, J.; Lotsch, B. V.; Schnick, W..2011,, 3213. doi: 10.1002/chem.201002462
(26) Zhao, J. N.; Ma, L.; Wang, H. Y.; Zhao, Y. F.; Zhang, J.; Hu, S. Z.. 2015,, 625. doi: 10.1016/j.apsusc.2015.01.233
(27) Li, S. J.; Chen, X.; Hu, S. Z.; Li, Q.; Bai, J.; Wang, F.. 2016,, 45931. doi: 10.1039/C6RA08817A
(28) Babu, G. A.; Ravi, G.; Mahalingam, T.; Kumaresavanji, M.; Hayakawa, Y..2015,, 4485. doi: 10.1039/C4DT03483J
(29) Schwenke, A. M.; Hoeppener, S.; Schubert, U. S.2015,, 23778. doi: 10.1039/C5TA06937H
(30) Dom, R.; Subasri, R.; Hebalkar, N. Y.; Chary, A. S.; Borse, P. H.. 2012,, 12782. doi: 10.1039/C2RA21910G
(31) Yuan, Y. P.; Yin, L. S.; Cao, S. W.; Gu, L. N.; Xu, G. S.; Du, P.; Chai, H.; Liao, Y. S.; Xue, C.. 2014,, 4663. doi: 10.1039/C4GC01517G
(32) Ding, Y.; Zhao, W.; Hu, H.; Ma, B. C..2008,, 910. doi: 10.1039/B808404A
(33) Saha, M.; Das, M.; Nasani, R.; Choudhuri, I.; Yousufuddin, M.; Nayek, H. P.; Shaikh, M. M.; Pathak, B.; Mukhopadhyay, S.. 2015,, 20154. doi: 10.1039/C5DT01471A
(34) Choi, J.; Zhang, S. H.; Hill, J. M.. 2012,, 179. doi: 10.1039/C1CY00301A
(35) Ding, Y. D.; Song, G.; Zhu, X.; Chen, R.; Liao, Q.. 2015,, 30929. doi: 10.1039/C4RA15127E
(36) Gu, Z. H.; Li, K. Z.; Qing, S.; Zhu, X.; Wei, Y. G.; Li, Y. T.; Wang, H.. 2014,, 47191. doi: 10.1039/C4RA06715K
(37) Kim, Y. I.; Atherton, S. J.; Brigham, E. S.; Mallouk, T. E.. 1993,, 11802. doi: 10.1021/j100147a038
(38) Kannapu, H. P. R.; Neeli, C. K. P.; Rao, K. S. R.; Kalevaru, V. N.; Martin, A.; Burri, D. R.. 2016,, 5494. doi: 10.1039/C6CY00397D
(39) Lee, S. W.; Heo, J.; Gordon, R. G.2013,, 8940. doi: 10.1039/c3nr03082b
(40) Zhou, X. S.; Jin, B.; Chen, R. Q.; Peng, F.; Fang, Y. P.. 2013,, 1447. doi:10.1016/j.materresbull.2012.12.038
(41) Xu, H.; Yan, J.; She, X. J.; Xu, L.; Xia, J. X.; Xu, Y. G.; Song, Y. H.; Huang, L. Y.; Li, H. M.2014,, 1406. doi: 10.1039/C3NR04759H
(42) Li, K. X.; Yan, L. S.; Zeng, Z. X.; Luo, S. L.; Luo; X. B.; Liu, X. M.; Guo, H. Q.; Guo, Y. H.. 2014,, 141. doi:10.1016/j.apcatb.2014.03.010
(43) Niu, P.; Yang, Y. Q.; Yu, J. C.; Liu, G.; Cheng, H. M..2014,, 10837. doi:10.1039/c4cc03060e
(44) Ge, L.; Han, C.. 2012,,268.doi: 10.1016/j.apcatb.2012.01.021
(45) Zhang, Y. W.; Liu, J. H.; Wu, G.; Chen, W.2012,, 5300. doi: 10.1039/c2nr30948c
(46) Xu, Y.; Xu, H.; Wang, L.; Yan, J.; Li, H.; Song, Y.; Huang, L.; Cai, G.. 2013,, 7604. doi: 10.1039/c3dt32871f
(47) He, B. L.; Dong, B.; Li, H. L.. 2007,, 425. doi: 10.1016/j.mseb.2007.06.017
(48) Huang, Q. W.;Tian, S. Q.; Zeng, D. W.; Wang, X. X.; Song, W. L.;Li, Y. Y.;Xiao, W.; Xie, C. S.. 2013,, 1477. doi: 10.1021/cs400080w
(49) Teranishi, M.; Naya, S.; Tada, H..2010,, 7850. doi: 10.1021/ja102651g
(50) Maurino, V.; Minero, C.; Mariella, G.; Pelizzetti, E.. 2005,, 2627. doi: 10.1039/b418789j
(51) Liu, G.; Niu, P.; Yin, L. C.; Cheng, H. M.. 2012,, 9070. doi: 10.1021/ja302897b
(52) Kim,H.; Kwon,O. S.; Kim,S.; Choi,W.; Kim, J. H.2016,, 1063.doi: 10.1039/c5ee03115j
熔盐辅助微波法制备g-C3N4包覆MgO-Al2O3-Fe2O3异质结催化剂及其光催化制过氧化氢性能
陈 鑫 胡绍争*李 萍 李 薇 马宏飞 陆 光
(辽宁石油化工大学化学化工与环境学部,辽宁 抚顺 113001)
工业上,双氧水的生产采用的是蒽醌法。此方法采用多步加氢和氧化过程,因此能耗很大。光催化制过氧化氢技术作为可持续和环境友好的新工艺,是传统蒽醌和电化学法的优秀替代者。本文采用熔盐辅助微波法制备了g-C3N4包覆MgO-Al2O3-Fe2O3异质结催化剂。制备的异质结催化剂在可见光下表现出优异的光催化制过氧化氢性能。熔盐的引入改变催化剂形貌的同时也影响了原料三聚氰胺的缩聚度,进而影响了其能带结构。制备的包覆结构能使两组分形成最大面积的异质结和强相互作用。这种强相互作用有利于光生电子-空穴对的分离和界面迁移,进而提高了过氧化氢的生成速率。制备的异质结催化剂的双氧水平衡浓度和生成速率分别为6.3 mmol·L−1和1.42 mmol·L−1·h−1,远高于两个单组份。不仅如此,制备的异质结催化剂还能抑制过氧化氢的分解。本文通过自由基捕获实验探讨了可能的反应机理和电子转移路径。
石墨相氮化碳;包覆结构;异质结;制过氧化氢;熔盐辅助微波法
O643
10.3866/PKU.WHXB201706153
May 9, 2017;
June 13, 2017;
June 15, 2017.
Corresponding author. Email: hushaozhenglnpu@163.com; Tel: +86-13470570415.
The project was supported by the National Natural Science Foundation of China (41571464), Education Department of Liaoning Province, China (L2014145), and Natural Science Foundation of Liaoning Province, China (201602467).
国家自然科学基金(41571464),辽宁省教育厅项目(L2014145)及辽宁省自然科学基金项目(201602467)资助
猜你喜欢
杂志排行

物理化学学报的其它文章
- AlN-Fe纳米复合薄膜:一种新型锂离子电池负极材料
- Theoretical and Experimental Studies on the Crystal Morphologyof Transition-Metal Carbohydrazide Perchlorate Complexes
- 无定型钼硫化物/还原氧化石墨烯的辐射合成及其电催化析氢性能
- Synthesis of Poly(bis-3,4-ethylenedioxythiophene methine)s with Side-Chain Comprising Electro-Optical Moieties and Alkyl Chain Effect in Solid State Polymerization
- 介孔TiO2薄膜光波导共振传感器对苯并(a)芘的探测灵敏度
- 量子点与蛋白质相互作用热力学