一种基于基因组甲基化探针的肺腺癌诊断模型
2018-01-10窦亚光田卫东
窦亚光,田卫东
(复旦大学 生命科学学院 生物统计学与计算生物学系,上海200438)
一种基于基因组甲基化探针的肺腺癌诊断模型
窦亚光,田卫东
(复旦大学 生命科学学院 生物统计学与计算生物学系,上海200438)
由于肺腺癌早期病征不够明显,传统的检测方法难以达到早期临床诊断的要求.目前,基于甲基化分子标记进行癌症的早期诊断则展现出很好的发展前景.本研究在训练集中筛选出在肺腺癌与正常样本中甲基化差异度最大的10个甲基化探针,并基于此构建广义线性诊断模型,同时引入Lasso方法进行模型的变量选择.最终得到由4个探针(分别对应于基因TRIM58、HOXA9、HOXB4、PRAC)作为变量的诊断模型,并提供了合理的分类阈值区间.在3个测试集使用该模型都表现出很好的诊断效果,ROC曲线的AUC均在0.99以上.
肺腺癌; 早期诊断; 甲基化; 探针; 广义线性模型; Lasso
如今,癌症是中国人死亡的首要原因,也是一个重要的公共卫生问题,其发病率和死亡率都在逐年增高[1].癌症按照患病部位分为多种,如肺癌、胃癌、肝癌等.其中,肺癌的发病人数和死亡人数最多,分别占所有癌症种类的18%与22%[2].据估计,2015年中国新增70多万肺癌病人,有60多万人死于肺癌[2].在肺癌中,非小细胞肺癌是其主要亚型,占到肺癌的85%左右(小细胞肺癌占到15%左右)[3].在现有的治疗水平下,非小细胞肺癌患者的总体预后水平差,患者在Ⅲ期和Ⅳ期等中晚期阶段对应的5年生存率分别为5%~14%和1%,但在癌症早期阶段诊治可以将生存率提高到50%[4],且相比于小细胞肺癌,非小细胞肺癌的增长较慢,在早期得到诊断并进行手术治疗能够根除[5].因此,对非小细胞肺癌的早期诊断是提高病人存活率,减少中国癌症死亡人数的直接而有效的途径.肺腺癌(Lung Adenocarcinoma, LUAD)是非小细胞肺癌最主要的亚型,占非小细胞肺癌的40%[6],且其发病率在许多国家呈不断上升趋势[7],是亟待深入研究的肺癌亚型.
以往的肺腺癌检测手段主要有胸部X光、痰细胞检测、光纤检查还有CT检查等检测手段,由于早期诊断率低等问题,未能有效降低肺腺癌患者的死亡率,这些检测手段在早期诊断时存在灵敏度低、错误率高、效率低下等缺点[8-9].再加上肺腺癌早期症状不够明显,超过半数的肺腺癌患者在确诊时已经发生了癌症转移[10-11].
过去十年来,利用生物分子对肺腺癌诊断的技术有了很大的进展,但是利用SNP、mRNA、microRNA和蛋白质来对肺腺癌等非小细胞肺癌进行诊断的效果还远达不到临床应用的水平(要求灵敏度和特异度都要在90%以上)[12-13].而DNA甲基化则展现出良好的发展前景.DNA甲基化发生在胞嘧啶鸟嘌呤二核苷酸(CpGs)上[14],在基因的表达调控与信使RNA可变剪接中起到重要功能.在出现时间方面,动物和人类的研究表明,基因甲基化的变化发生在肺癌癌变的早期,并存在于肺癌的癌前病变中[15-16].在甲基化差异特征方面,位于肿瘤抑制基因启动子区域和管家基因的CpG岛甲基化水平的提高以及基因组全局脱甲基化都是癌细胞的常见特征[17-18].在技术方面,高通量甲基化芯片现在已经可用于同时测定数千个CpG位点的甲基化水平[19],且DNA甲基化具有稳定、易定性或定量检测的优点.在样本获取途径方面,肺腺癌患者中的癌组织、血液、痰等多种样本中都可以定量检测到这样的差异甲基化数据[20].综合上述多方面优势,DNA甲基化已逐渐成为癌症早期检测的最有希望的诊断标记物之一[21],并且找到合适的用于癌症诊断的甲基化位点或基因组合已经成为现在研究的重点[19,22].
基于高通量甲基化数据,本研究综合利用多个差异甲基化位点来构建肺腺癌的诊断模型.本研究引入了Lasso方法进行模型构建,Lasso方法可在保持模型准确率的前提下进行变量筛选与模型精简[23].我们在构建模型的同时引入Lasso方法来对基因甲基化位点进行筛选,利用筛选出的甲基化位点构建广义线性模型.最后,我们利用独立的测试集对模型的诊断效果进行评估.
1 数据与方法
1.1 数据搜集
本研究收集了来自同一甲基化平台(Human Methylation27 DNA Analysis BeadChip)的4组肺腺癌level3的甲基化芯片数据.其中3组下载自GEO数据库[24](http:∥www.ncbi.nlm.nih.gov/geo/),对应的GEO记录号分别为GSE32861(59个正常样本,59个癌症样本),GSE62948(28个正常样本,28个癌症样本),GSE32866(27个正常样本,28个癌症样本).一组下载自TCGA数据库[25](http:∥portal.gdc.cancer.gov/),包含24个正常样本,126个癌症样本.数据分析平台对应的GEO平台编号为GPL8490,利用该芯片平台可测量14495个人类基因上27578个CpG位点的甲基化水平.本研究以GSE32861对应的数据为训练集,剩余其他3组数据作为独立测试集.
同时收集了TCGA中对应Human Methylation27 DNA Analysis BeadChip平台的所有癌症患者正常样本的甲基化数据,共计360个.这些数据分别对应10种癌症,分别为: 肺腺癌,肺鳞状细胞癌(Lung Squamous Cell Carcinoma, LUSC),子宫内膜癌(Uterine Corpus Endometrial Carcinoma, UCEC),胃腺癌(Stomach Adenocarcinoma, STAD),肾乳头状细胞癌(Kidney Renal Papillary Cell Carcinoma, KIRP),肾透明细胞癌(Kidney Renal Clear Cell Carcinoma, KIRC),卵巢浆液性囊腺癌(Ovarian Serous Cystadenocarcinoma, OV),乳腺浸润性癌(Breast Invasive Carcinoma,BRCA),结肠腺癌(Colon Adenocarcinoma,COAD),直肠腺癌(Rectum Adenocarcinoma, READ).其对应的正常样本数目见表1.将这360个不同癌症的正常样本数据与126个TCGA平台的肺腺癌癌症样本数据合并,作为混合数据集.
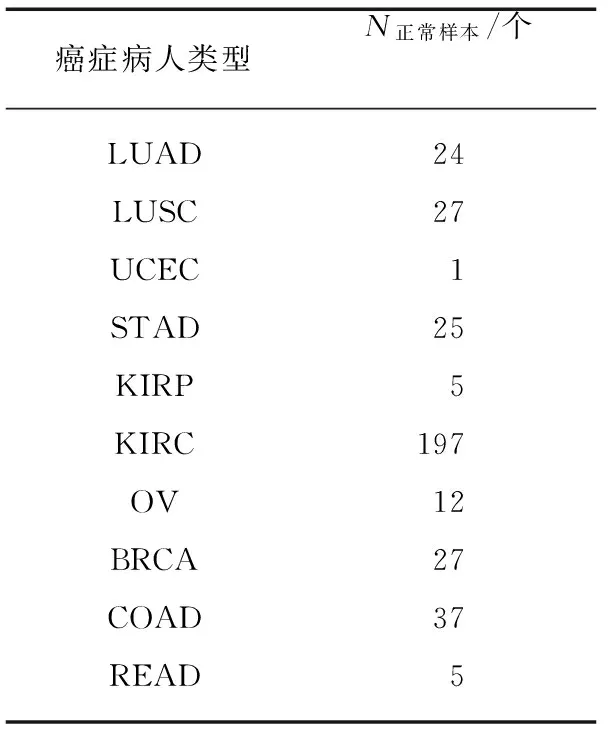
表1 不同癌症病人的正常样本数目Tab.1 Normal sample sizes of different cancer patients
甲基化芯片level3记录的是CpG位点的β值,计算公式为β=M/(M+U+α),其中M为该位点的甲基化信号强度,U为非甲基化信号强度,α是常数项,默认为100[26].β可以被用来度量基因组CpG位点的甲基化水平[26].
1.2 甲基化数据的预处理
首先去除所有定位在性染色体(X和Y染色体)的探针,只考虑常染色体对应的探针位点,保证本研究构建的模型的通用性.为了获取高质量的探针位点,删除了在不同甲基化样本中存在空值的探针.为了去除每组甲基化数据内部的技术误差(technical variation),利用分位数正则化的方法对每组甲基化数据进行数据集内部校正,该操作通过R语言中的lumi包[27]来实现.为了去除不同数据批次之间的系统误差,利用经验贝叶斯方法对4组甲基化数据进行批次间校正[28],该处理过程通过R语言SVA包[29]中的ComBat函数来完成.
1.3 差异甲基化的计算
对于每一个探针,本研究采用5%分位数、25%分位数、50%分位数、75%分位数、95%分位数这5个数值来表征该探针位点在样本中甲基化水平(β值)的分布.然后计算探针在癌症样本与正常样本中对应的5个分位数值差值的加和,利用该加和的绝对值的大小来指代该探针位点在癌症样本与正常样本中的甲基化差异度.
1.4 模型构建与评估
利用1.3节计算探针位点在癌症与正常样本中甲基化差异度的方法,挑选出甲基化差异度排名在前10的探针作为模型构建的候选探针.基于广义线性模型构建肺腺癌诊断模型.模型中以样本中选出的探针对应的甲基化β值作为候选输入变量,输出结果为一个得分,得分越高代表诊断该样本为肺腺癌癌症样本的可能性越大.为了对探针变量进行进一步筛选并防止模型的过拟合,在模型的训练过程中引入Lasso[23]方法来对变量系数进行惩罚,从而在保证模型诊断效果的同时实现模型变量的精简[23].广义线性模型的训练与参数的训练通过R语言glmnet包[30]中的cv.glmnet函数来实现.该函数通过训练集内部的十倍交叉验证过程来搜索最佳的Lasso惩罚权重,并得到在该惩罚权重下不同变量的参数取值.
通过上述模型参数的筛选与训练,得到在广义线性模型下探针的变量选取结果与不同探针参数的取值.并测试构建好的广义线性模型在其他3组独立的测试集中区分癌症与正常样本的表现.将测试集样本中对应变量探针的β值作为输入,输入广义线性模型计算输出诊断得分,并与测试数据集本身的样本类型标签进行对应.诊断结果中,被诊断样本本身为癌症样本且被诊断为癌症样本则该类诊断结果为真阳性结果(TP),被诊断样本本身为癌症样本但被诊断为正常样本则该类诊断结果为假阴性结果(FN),被诊断样本本身为正常样本且被诊断为正常样本则该类诊断结果为真阴性结果(TN),被诊断样本本身为正常样本但被诊断为癌症样本则该类诊断结果为假阳性结果(FP).然后分别采用特异度(Specificity=TN/(TN+FP)),灵敏度(Sensitivity=TP/(TP+FN)),AUC(ROC曲线下方面积),AUCPR(Precision-recall曲线下方面积)来评估模型的肺腺癌诊断能力.上述4种评估方式的取值都在0到1之间,数值越大代表模型的区分效果越好.
2 结果与分析
2.1 探针的挑选与质量控制
具体流程图见图1.挑选出所有定位在常染色体上的探针,并删除在不同甲基化样本中存在空值的探针,这样一共得到了19626个甲基化探针.对4组数据进行数据集内与数据间的校正.然后利用筛选校正之后的甲基化数据通过1.3节所述方法计算得到这些探针位点在癌症样本与正常样本中的甲基化差异度.
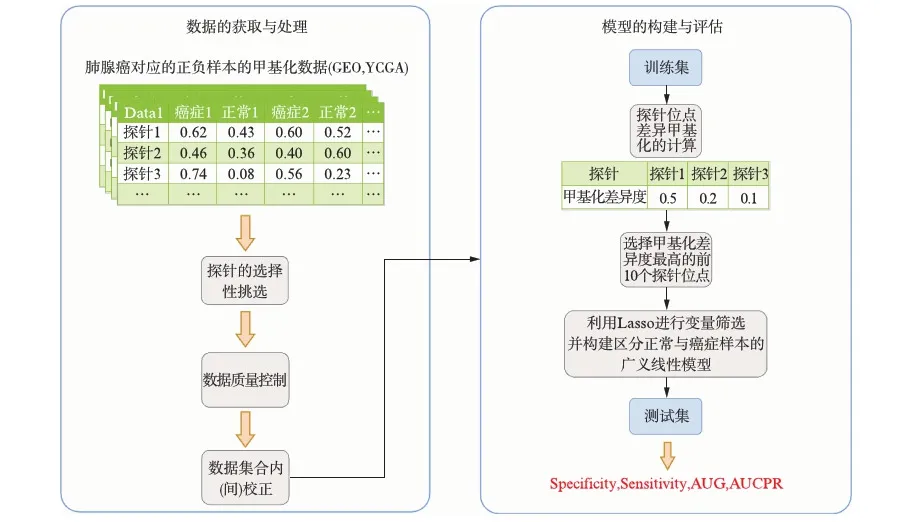
图1 数据的获取、处理与模型的构建评估流程图Fig.1 Flowchart of data collection and processing as well as the model building and performance evaluation
2.2甲基化的差异分析
按照甲基化差异度对探针进行排序,选取排名位于前10的探针,作为候选变量探针以构建肺腺癌诊断模型,它们分别为cg12374721、cg26521404、cg25720804、cg08089301、cg14458834、cg07533148、cg00949442、cg23432345、cg22881914、cg23290344,对应的基因分别为PRAC、HOXA9、TLX3、HOXB4、HOXB4、TRIM58、ABCA3、HOXA7、NID2、NEF3.这些探针在癌症样本中的甲基化水平均值都是其在正常样本中的2.5倍以上,它们在癌症与正常样本中的具体分布情况见图2.这些探针都在癌症中呈现高甲基化状态,在正常样本中呈现相对较低的甲基化状态,样本间甲基化差异程度大,具有一定的区分癌症样本与正常样本的能力.但可以看到对于单个甲基化位点来说,癌症与正常样本的甲基化分布区间存在一定程度的重合,仅靠单个甲基化位点对癌症与正常样本进行区分,其效果存在局限性.合理地综合利用上述探针,挑选探针组合,构建癌症与正常样本的区分模型,从而克服单甲基化位点诊断的局限性,以提高模型对癌症与正常样本的区分效果.
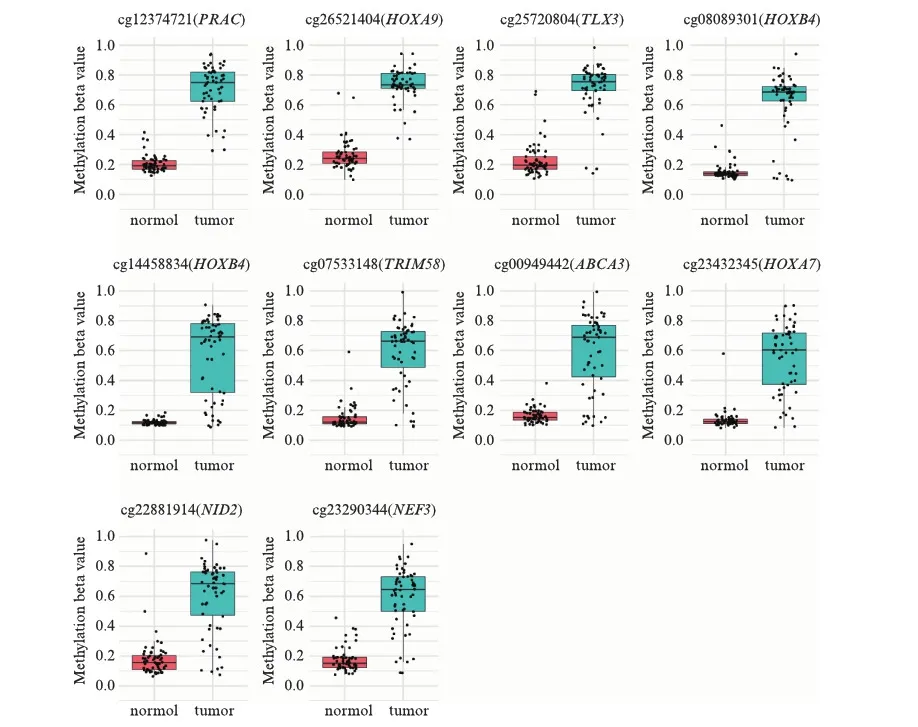
图2 甲基化差异度处在前10名的探针在正常与癌症样本中的甲基化水平分布Fig.2 Distribution of the methylation levels of top 10 probes ranked by methylation difference in normal and tumor samples
2.3 模型的构建与评估
利用GSE32861数据(59个正常样本,59个癌症样本)作为训练数据集,来构建用来诊断肺腺癌的广义线性诊断模型.构建模型的方式是: 利用cv.glmnet[30]工具并引入Lasso方法进行广义线性模型的训练,对候选探针进行筛选后,选取了10个探针中的4个探针cg07533148、cg08089301、cg12374721、cg26521404作为有效模型变量.对应的基因分别为TRIM58、HOXB4、PRAC、HOXA9.模型探针具体的变量参数见表2.
之后,利用3组独立测试集对构建的广义线性模型的性能进行评估.评估结果如表3所示,在不同的诊断集合中,分别评估当特异度为100%时对应的灵敏度数值,也就是所有的正常样本都被正确分类的情况下评估模型能诊断出多少比例的癌症样本.可以看到在训练集(GSE32861)中当特异度为100%时,对应的灵敏度为98.3%,在3组测试集中灵敏度都在96%以上,尤其是在GSE62948数据集中,灵敏度达到了100%,也就是在该数据集中模型可以做到样本类别的全部正确区分.该模型在所有数据集中的AUC与AUCPR值都在0.99以上.以上结果一定程度上说明了该模型具有良好的区分癌症样本与正常样本的能力.
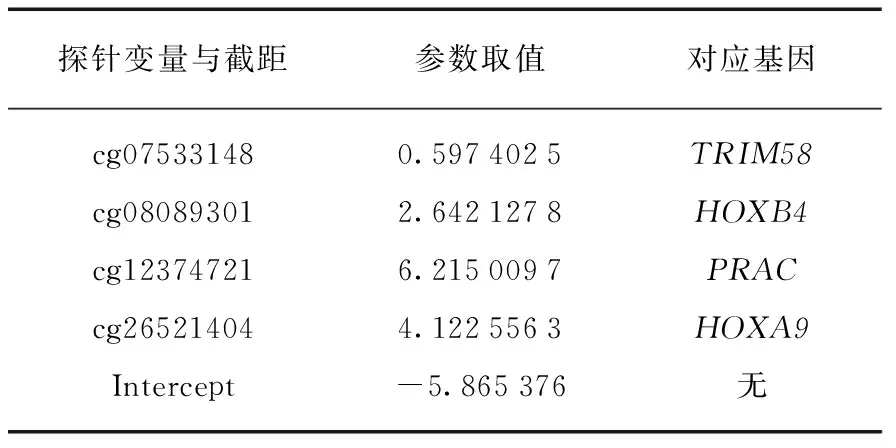
表2 模型探针变量与参数取值Tab.2 Coefficients of probe variables in the model
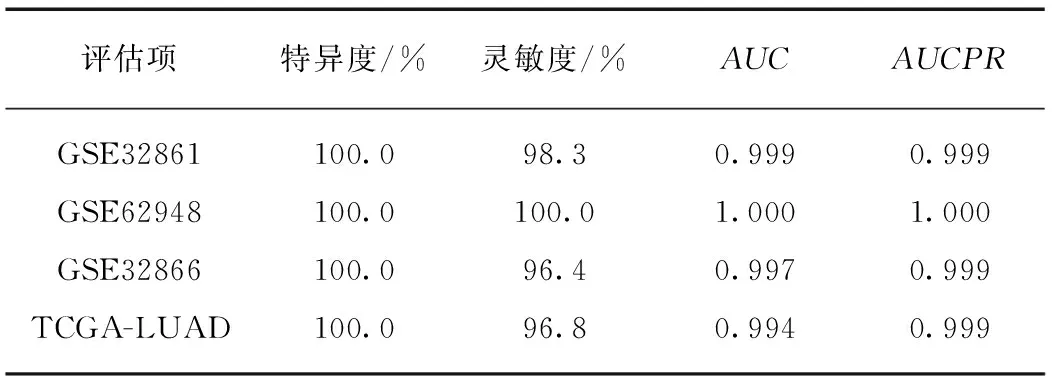
表3 模型在训练集与测试集中的表现Tab.3 Performance of the model in training and testing datasets
广义线性模型对每个输入样本都有一个评估得分,分值越高则代表越可能被诊断为癌症样本,基于该模型不同数据集正常样本与癌症样本所对应的得分分布如图3所示,其中下标为ALL的一组数据指代的是将4个数据集的所有样本进行合并之后的数据集.可见在不同数据集上,正常样本与癌症样本对应的模型得分都有很大差异,且正常样本对应的分值都集中在较低的水平,绝大多数都在0.15以下,对于TCGA-LUAD数据集对应癌症样本来说,其得分的分布则相较于其他2个数据集较为分散,但其大部分的癌症样本分值仍在0.15以上.在利用模型进行诊断时,会设置一个分类阈值,样本得分大于阈值时则被诊断为癌症样本,样本得分小于阈值时则被诊断为正常样本,不同的阈值选取可能会产生不同的模型诊断结果.
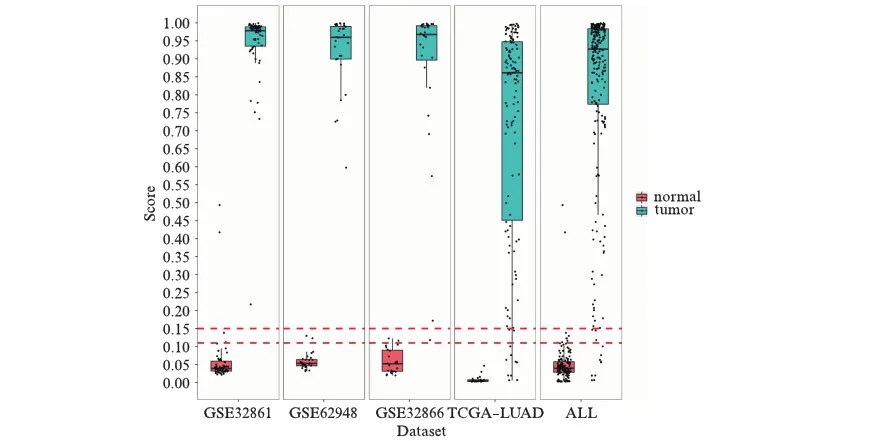
图3 不同数据集正常样本与癌症样本对应的模型分值分布图Fig.3 The model-generated score distribution of normal and tumor samples from different datasets
在保证模型在所有的数据集上的特异度与灵敏度都在90%及其以上的情况下,对于每一个数据集都可以得到一个分类阈值区间,GSE32861、GSE62948、GSE32866还有TCGA-LUAD这4个数据集对应的阈值区间分别为0.1~0.83、0.09~0.72、0.11~0.57、0.02~0.15.这4个数据集的阈值区间交集为0.11~0.15,如图3中红色虚线所标注,也就是当所选分类阈值在该区间内时,利用构建的模型在4个数据集上进行诊断分类,对应的特异度与灵敏度都在90%及其以上.为了评估在阈值交集区间上模型的诊断区分效果,选择0.11与0.15的平均值0.13作为分类阈值以考察模型诊断性能,此时4个数据集对应的特异度分别为94.9%、100%、100%、100%,对应的灵敏度分别为100%、100%、96.4%、92.9%.在该分类阈值下,GSE62948与GSE32866均可以达到当特异度为100%时的理想水平.但对于TCGA-LUAD与GSE32861这两个数据集的诊断效果则未达到理想水平,对于TCGA-LUAD数据集来说,其灵敏度相较于96.8%有所下降,对于数据集GSE32861来说,当其灵敏度为100%时的最优特异度为96.6%,高于在该分类阈值下的94.9%.因此本研究建议,若存在合适的历史数据,利用数据对模型的阈值进行进一步训练选择则可以一定程度上改善模型的诊断性能,获取更好的诊断效果.在使用该模型进行癌症诊断时,若不存在历史数据,则可以采用0.11~0.15区间内的数值(例如0.13)作为模型输出值的分类阈值直接进行诊断使用.
2.4 模型对不同癌症分期癌症样本的诊断效果
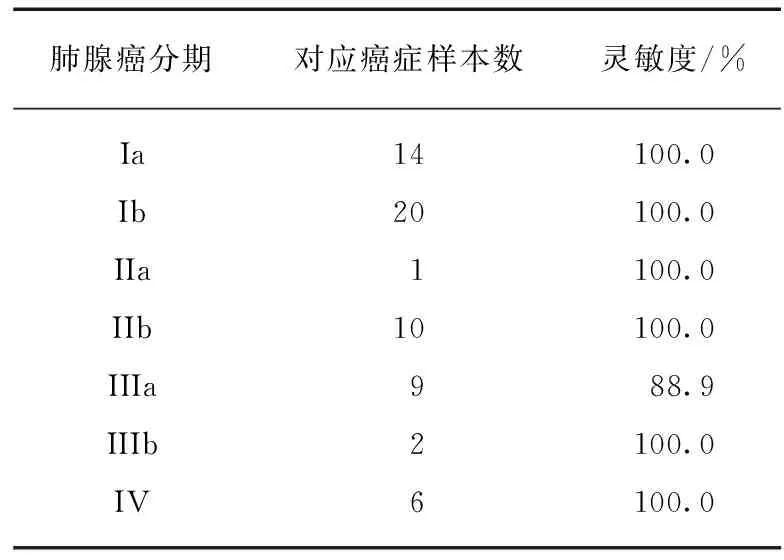
表4 不同分期癌症样本诊断灵敏度Tab.4 Diagnostic sensitivity of tumor samples for different tumor stages
TCGA-LUAD数据集的126个癌症样本中有62个样本存在癌症阶段数据,一共对应癌症的Ia、Ib、IIa、IIb、IIIa、IIIb、IV等7个时期.利用0.13作为模型分类阈值时,模型在TCGA-LUAD数据集上对应的特异度为100%,即此时所有的正常样本均被正确分类,然后考察此时不同时期的癌症样本数目与其对应的灵敏度,如表4所示.对于Ia、Ib这两个相对较早的癌症时期来说,灵敏度达到了100%,也就是所有标记为该时期的癌症样本都被正确诊断,对于IIa、IIb、IIIb、IV等4个时期也同样如此,9个IIIa期的癌症样本也有8个被成功诊断.说明了模型在对癌症进行诊断区分时,对于不同时期的癌症样本均能取得良好的诊断效果.对于Ia、Ib两类相对早期的癌症样本,其良好的诊断效果一定程度上说明了利用该模型进行肺腺癌早期诊断的可行性.由于数据样本量较少,未来更多的癌症样本分期数据的加入则可以更加精确地评估模型对不同癌症分期样本的诊断性能.
2.5 模型在混合数据集上的性能评估
为了进一步评估模型区分不同类型癌症病人的正常样本与肺腺癌癌症样本的能力.利用构建好的模型对混合数据集进行正常样本与癌症样本的区分,此时对应的AUC与AUCPR分别为0.978与0.943,诊断结果的ROC曲线如图4(a)所示.当约登指数(Youden’s index)也就是Sensitivity+Specificity-1取值最大时,对应的特异度与灵敏度分别为90.0%与92.9%,此时对应的分类阈值为0.122,在上述的0.11~0.15的区间之内.虽然此时模型的诊断表现相较于前2个独立测试集有一定程度的下降,但特异度与灵敏度都仍保持在90%及其以上水平.除了结肠腺癌、胃腺癌还有卵巢浆液性囊腺癌外,对于混合数据集中其余7种类型癌症对应的正常样本均能做到完全正确区分,也就是对应的特异度都是100%,具体如图4(b)所示.以上结果说明在样本类型状态未知时,利用本研究构建的模型区分不同人体部位或类型的正常样本与肺腺癌癌症样本,在一定程度上也具有良好的诊断性能与应用前景.
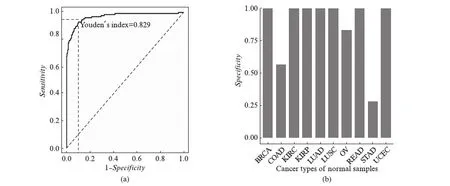
图4 利用模型对混合数据集进行诊断时的ROC曲线图(a)与不同癌症患者正常样本的特异度图(b)Fig.4 The ROC curve(a) of diagnostic performance when applying the model to mixed dataset and the specificity chart(b) of normal samples from different cancer patients
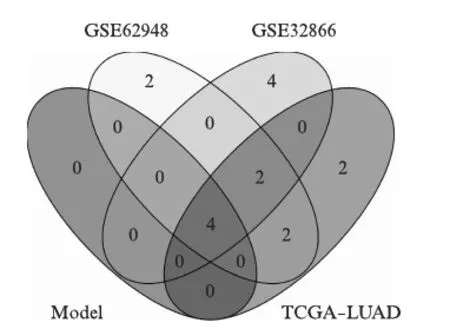
图5 3个测试集甲基化差异度排名前10探针 与4个模型变量探针重合图Fig.5 Overlap details of the top 10 probes ranked by methylation difference in three test datasets and four probes included in the model
2.6 模型所使用探针的差异甲基化排名一致靠前
为了进一步研究所选探针的特征,我们计算了19626个探针在3个测试集中的甲基化差异度,分别在每个数据集中按照甲基化差异度从高到低对探针进行排序.取各组数据中排名前10的探针与模型使用的4个变量探针,观察它们的重合情况.详情见图5,可以看到4个模型变量探针无一例外都在3个数据集的前10名中出现.从4个探针在不同数据集的具体排名来看,4个探针在不同的数据集中的排名的均值都在6以内.这些在一定程度上说明了模型使用的探针在不同数据集中差异甲基化排名的稳定性,整体上在不同数据集中都处于排名靠前的位置,这也为该模型的推广应用提供了一定程度的支持.
2.7 所选探针的文献验证
为了探究模型所选取的探针对应基因与肺腺癌的关联意义,本研究对它们分别进行了文献验证.在本研究中,这些探针都在肺腺癌中处于高甲基化状态,它们对应的基因分别为TRIM58、HOXA9、HOXB4、PRAC.其中TRIM58、HOXA9、HOXB4在肺腺癌中的高甲基化现象在已有文献中均存在相关报道.本研究中,位于TRIM58基因的探针在癌症样本中表现为高甲基化,其甲基化水平均值约为正常样本的4.7倍.以往研究表明,TRIM58在肺腺癌的早期就被稳定地高甲基化,是肺腺癌中最突出的候选抑癌基因之一,该基因的过表达可以在体内和体外抑制肺腺癌肿瘤的生成与增殖[31].本研究中,位于HOXA9基因的探针在癌症样本中表现为高甲基化,其甲基化水平均值约为正常样本的2.7倍.Hulbert等通过对肺癌患者和正常人的痰和血液进行基因甲基化检测,发现基因HOXA9在肺癌的早期出现显著的高甲基化现象,并认为可以将HOXA9作为潜在的甲基化标记物用于癌症的早期诊断[32],与本研究一致.HOXA9是7号染色体上homebox A基因簇的一员,编码与DNA结合的转录因子,存在于KEGG[33]的癌症中转录失调(transcriptional misregulation in cancer)通路中,具有潜在的调控基因表达与细胞分化的功能[34],并参与肺癌细胞的恶化转移过程,在肺癌细胞的攻击性方面起到潜在的核心调控功能[35].HOXA9基因不仅在肺腺癌中存在高甲基化现象,在嗜酸细胞瘤[36]、脑膜瘤[37]、膀胱癌[38]、口腔癌[39]中也存在着显著的差异甲基化现象,并被报道可作为这些癌症诊断的潜在分子标记物.在本研究中,位于HOXB4基因的探针在癌症样本中表现为高甲基化,其甲基化水平均值约为正常样本的4.0倍.Daugaard等在2016年的研究中指出HOXB4基因在肺腺癌组织中存在显著的高甲基化现象,可以作为肺腺癌诊断的潜在甲基化分子标记物[40],与本研究的结论一致.HOXB4基因在非小细胞肺癌中的高甲基化与癌细胞的迁移与转移存在密切关联[41].同时HOXB4基因在癌症中的高甲基化也出现在急性髓性白血病[42]、口腔癌[39]、甲状腺癌[14]、肝外胆管癌[43]等癌症中.
3 讨 论
随着对癌症早期基因差异甲基化研究的深入,发现基因的甲基化特征与癌症的患病[5]、分型[44]、预后[19]、复发[45]等都存在关联.基因的差异甲基化在癌症早期就会出现[15-16],甲基化芯片为甲基化的测定提供了技术便利[19],利用差异甲基化对癌症进行早期诊断拥有很好的发展前景与应用价值.
本研究对候选探针按照其在癌症样本与正常样本中的甲基化的差异度排序,利用Lasso[23]方法对前10名的探针进行进一步的变量挑选,最终选出其中4个探针构建出了区分癌症样本与正常样本的广义线性模型.并在多个数据集中对模型的区分能力进行了评估,对应的AUC值都在0.99以上,具有良好的癌症样本正常样本区分效果.并且对癌症早期的病人诊断时在有限数据内特异度与灵敏度都达到100%,说明了利用本研究构建模型进行肺腺癌早期诊断的可行性与应用价值.同时给出了模型在使用时分类阈值取值的合理区间(0.11~0.15),以供实现在没有训练数据情况下的诊断之用.这4个探针在不同数据集中的甲基化差异程度排名名次都处于前10名以内,其甲基化差异度的排名都一致靠前,一定程度上说明利用这些探针对肺腺癌进行前期诊断推广应用的潜力.
模型所选的4个候选探针分别对应于基因TRIM58、HOXA9、HOXB4和PRAC.其中TRIM58、HOXA9、HOXB4的高甲基化状态与肺腺癌密切相关.值得注意的是,HOXA9与HOXB4不仅在肺腺癌中存在高甲基化状态,分别在其他如脑膜瘤[37]、口腔癌[39]等多种癌症中也存在明显的基因差异甲基化现象.这在一定程度上说明利用甲基化探针集合同时对多个可能癌症进行检测诊断存在潜在可能.在对混合数据集进行诊断时,对结肠腺癌、胃腺癌还有卵巢浆液性囊腺癌的正常样本的区分度还不够,未来逐渐获取更广泛的有关不同癌症的甲基化检测数据,寻找组织与癌症特异性的甲基化分子标记,在对特定的癌症进行精确的诊断分析并排除其他组织的干扰,以进行精确的癌症检测与癌症分类,不断精进基于甲基化进行癌症诊断,则是一个未来发展的方向.同时本研究还发现了PRAC基因对应的cg12374721探针在模型中具有最高的变量系数且具有很高的甲基化差异排名,但未见于已有的报道中,该探针以及对应的基因或许可以作为新的潜在的肺腺癌诊断标记物.以往的多数对肺腺癌与正常组织的基因甲基化差异研究以及本研究,都旨在寻找肺腺癌诊断的潜在标记物或设计诊断探针组合,但缺乏对所选分子标记物甲基化与肺腺癌功能机制方面的了解与深入探究,在寻找肺腺癌诊断标记物的同时对这方面的分子功能机制的揭示或许可以成为后续研究的一个方向.
[1] MA J T. China statistical yearbook [R]. Beijing: National Bureau of Statistics of China, 2010.
[2] CHEN W, ZHENG R, BAADE P D,etal. Cancer statistics in China, 2015 [J].CA:ACancerJournalforClinicians, 2016,66(2): 115-132.
[3] ATLANTA G. Cancer facts and figures 2017[EB/OL].AmericanCancerSociety, 2017. www.cancer.org/research.
[4] HANKEY B F, RIES L A, EDWARDS B K. The surveillance, epidemiology, and end results program [J].CancerEpidemiologyandPreventionBiomarkers, 1999,8(12): 1117-1121.
[5] LOKK K, VOODER T, KOLDE R,etal. Methylation markers of early-stage non-small cell lung cancer [J].PLoSOne, 2012,7(6): e39813.
[6] TRAVIS W D, T L B D S. Lung cancer [J].Cancer, 1995,75: 191-202.
[7] YOSHIMI I, OHSHIMA A, AJIKI W,etal. A comparison of trends in the incidence rate of lung cancer by histological type in the Osaka Cancer Registry, Japan and in the surveillance, epidemiology and end results program, USA [J].JapaneseJournalofClinicalOncology, 2003,33(2): 98-104.
[8] BACH P B, JETT J R, PASTORINO U,etal. Computed tomography screening and lung cancer outcomes [J].Jama, 2007,297(9): 953-961.
[9] International Early Lung Cancer Action Program Investigators. Survival of patients with stage I lung cancer detected on CT screening [J].NEnglJMed, 2006,2006(355): 1763-1771.
[10] SINGHAL S, VACHANI A, ANTIN-OZERKIS D,etal. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: A review [J].ClinicalCancerResearch, 2005,11(11): 3974-3986.
[11] WARDWELL N R, MASSION P P. Novel strategies for the early detection and prevention of lung cancer [J].SeminarsinOncology, 2005,32(3): 259-268.
[12] LI H, YANG L X, ZHAO X Y,etal. Prediction of lung cancer risk in a Chinese population using a multifactorial genetic model [J].BMCMedicalGenetics, 2012,13(1): 118.
[13] GUO S C, WANG Y L, LI Y,etal. Significant SNPs have limited prediction ability for thyroid cancer [J].CancerMed-Us, 2014,3(3): 731-735.
[14] BIRD A. DNA methylation patterns and epigenetic memory [J].Genes&Development, 2002,16(1): 6-21.
[15] BELINSKY S A, NIKULA K J, PALMISANO W A,etal. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis [J].ProcNatlAcadSciUSA, 1998,95(20): 11891-11896.
[16] ZOCHBAUER-MULLER S, MINNA J D, GAZDAR A F. Aberrant DNA methylation in lung cancer: biological and clinical implications [J].TheOncologist, 2002,7(5): 451-457.
[17] GRONBAEK K, HOTHER C, JONES P A. Epigenetic changes in cancer [J].Apmis, 2007,115(10): 1039-1059.
[18] TAKAI D, JONES P A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22 [J].ProcNatlAcadSciUSA, 2002,99(6): 3740-3745.
[19] KUO I Y, JEN J, HSU L H,etal. A prognostic predictor panel with DNA methylation biomarkers for early-stage lung adenocarcinoma in Asian and Caucasian populations [J].JournalofBiomedicalScience, 2016,23(1): 58.
[20] BELINSKY S A. Gene-promoter hypermethylation as a biomarker in lung cancer [J].NatRevCancer, 2004,4(9): 707-717.
[21] ZHAO Y X, SUN J F, ZHANG H Y,etal. High-frequency aberrantly methylated targets in pancreatic adenocarcinoma identified via global DNA methylation analysis using methylCap-seq [J].ClinicalEpigenetics, 2014,6(1): 18.
[22] TSOU J A, GALLER J S, SIEGMUND K D,etal. Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma [J].MolecularCancer, 2007,6(1): 70.
[23] TIBSHIRANI R. Regression shrinkage and selection via the Lasso: A retrospective [J].JRStatSocB, 2011,73(273-82).
[24] BARRETT T, TROUP D B, WILHITE S E,etal. NCBI GEO: Mining tens of millions of expression profiles-database and tools update [J].NucleicAcidsRes, 2007,35: 760-765.
[25] Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma [J].Nature, 2014,511(7511): 543.
[26] BIBIKOVA M, LIN Z W, ZHOU L X,etal. High-throughput DNA methylation profiling using universal bead arrays [J].GenomeRes, 2006,16(3): 383-393.
[27] DU P, KIBBE W A, LIN S M. Lumi: A pipeline for processing Illumina microarray [J].Bioinformatics, 2008,24(13): 1547-1548.
[28] JOHNSON W E, LI C, RABINOVIC A. Adjusting batch effects in microarray expression data using empirical Bayes methods [J].Biostatistics, 2007,8(1): 118-127.
[29] LEEK J T, STOREY J D. Capturing heterogeneity in gene expression studies by surrogate variable analysis [J].PLoSGenet, 2007,3(9): 1724-1735.
[30] FRIEDMAN J, HASTIE T, TIBSHIRANI R. Glmnet: Lasso and elastic-net regularized generalized linear models [J].RPackageVersion, 2009,1(4).
[31] KAJIURA K, MASUDA K, NARUTO T,etal. Frequent silencing of the candidate tumor suppressor TRIM58 by promoter methylation in early-stage lung adenocarcinoma [J].Oncotarget, 2017,8(2): 2890-2905.
[32] HULBERT A, JUSUE-TORRES I, STARK A,etal. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum [J].ClinicalCancerResearch, 2017,23(8): 1998-2005.
[33] KANEHISA M, GOTO S. KEGG: Kyotoencyclopediaof genes and genomes [J].NucleicAcidsRes, 2000,28(1): 27-30.
[34] HWANG S H, KIM K U, KIM J E,etal. Detection of HOXA9 gene methylation in tumor tissues and induced sputum samples from primary lung cancer patients [J].ClinChemLabMed, 2011,49(4): 699-704.
[35] YU S L, LEE D C, SOHN H A,etal. Homeobox A9 directly targeted by miR-196b regulates aggressiveness through nuclear Factor-kappa B activity in non-small cell lung cancer cells [J].MolCarcinogen, 2016,55(12): 1915-1926.
[36] PIRES-LUIS A S, COSTA-PINHEIRO P, FERREIRA M J,etal. Identification of clear cell renal cell carcinoma and oncocytoma using a three-gene promoter methylation panel [J].JournalofTranslationalMedicine, 2017,15(1): 149.
[37] GALANI V, LAMPRI E, VAROUKTSI A,etal. Genetic and epigenetic alterations in meningiomas [J].ClinNeurolNeurosur, 2017,158: 119-125.
[38] LOPEZ J I, ANGULO J C, MARTIN A,etal. A DNA hypermethylation profile reveals new potential biomarkers for the evaluation of prognosis in urothelial bladder cancer [J].APMIS, 2017,125(9): 787-796.
[39] XAVIER F C A, DESTRO M F D S, DUARTE C M E,etal. Epigenetic repression of HOXB cluster in oral cancer cell lines [J].ArchOralBiol, 2014,59(8): 783-789.
[40] DAUGAARD I, DOMINGUEZ D, KJELDSEN T E,etal. Identification and validation of candidate epigenetic biomarkers in lung adenocarcinoma [J].ScientificReports, 2016,6: 35807
[41] LIN S H, WANG J, SAINTIGNY P,etal. Genes suppressed by DNA methylation in non-small cell lung cancer reveal the epigenetics of epithelial-mesenchymal transition [J].BMCGenomics, 2014,15(1): 1079.
[42] QU X Y, DAVISON J, DU L,etal. Identification of differentially methylated markers among cytogenetic risk groups of acute myeloid leukemia [J].Epigenetics-Us, 2015,10(6): 526-535.
[43] SHU Y, WANG B, WANG J,etal. Identification of methylation profile of HOX genes in extrahepatic cholangiocarcinoma [J].WorldJGastroentero, 2011,17(29): 3407-3419.
[44] TOYOOKA S, MARUYAMA R, TOYOOKA K O,etal. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer [J].IntJCancer, 2003,103(2): 153-160.
[45] SELIGSON D B, HORVATH S, SHI T,etal. Global histone modification patterns predict risk of prostate cancer recurrence [J].Nature, 2005,435(7046): 1262-1266.
AModelforDiagnosisofLungAdenocarcinomaBasedonGeneMethylationProbe
DOUYaguang,TIANWeidong
(DepartmentofBiostatisticsandComputationalBiology,SchoolofLifeSciences,FudanUniversity,Shanghai200438,China)
Since there is lack of obvious symptoms in the early stage of lung adenocarcinoma, the traditional detection methods hardly meet the requirements of early clinical diagnosis. Currently, early detection of lung adenocarcinoma using DNA methylation biomarkers shows great promise. In this study, after analyzing of the training dataset including tumor and normal samples we chose the ten most differentially methylated probes. These ten probes are then used to build the general linear model to do lung adenocarcinoma diagnosis. It should be noted that, Lasso method is introduced in the model to perform variable selection. Finally, the lung adenocarcinoma diagnosis model is built based on the methylation level of four probes corresponding to four genes:TRIM58,HOXA9,HOXB4 andPRAC. And a reasonable classification score threshold interval is provided. The diagnosis performance of the model is pretty good when applying it to three independent test datasets, and theAUCsof all three ROC curves are greater than 0.99.
lung adenocarcinoma; early stage diagnosis; methylation; probe; general linear model; Lasso
0427-7104(2017)06-0671-10
2017-03-22
教育部博士点专项科研基金(博导类)(20120071110018)
窦亚光(1991—),男,硕士研究生;田卫东,男,教授,通信联系人,E-mail: weidong.tian@fudan.edu.cn.
Q332
A
