乙炔氢氯化超低固汞催化体系中钾对使用性能影响的机理研究
2018-01-03余延芳李武斌贵州省不动产登记中心贵州贵阳550000贵州银星集团贵州贵阳550000
余延芳,李武斌,张 彬(.贵州省不动产登记中心,贵州贵阳550000;.贵州银星集团,贵州贵阳550000)
乙炔氢氯化超低固汞催化体系中钾对使用性能影响的机理研究
余延芳1,李武斌2,张 彬2(1.贵州省不动产登记中心,贵州贵阳550000;2.贵州银星集团,贵州贵阳550000)
采用浸渍法制备HgCl2-KCl/C超低固汞催化剂,并使用固定床转化器对催化剂催化性能进行测试,用管式恒温炉对低汞催化剂的固汞效果进行评价。结果表明:KCl和HgCl2可以形成配合物K2HgCl4,增加了活性组分分散度,提高了催化活性,加强了固汞效果,其催化机理为络合催化。
超低固汞催化剂;机理;配合物;络合催化
当前无汞催化剂未能应用于工业化生产,超低固汞催化剂(氯化汞含量为4.0%)的研发对于PVC行业的发展及汞减排具有重要意义,本文以制备乙炔氢氯化的超低固汞催化剂为出发点,选用助剂氯化钾,对影响超低固汞催化剂使用性能的机理进行了分析。
1 试验
1.1 试验原料
活性炭填装密度:(430±20)g/L,四氯化碳吸附值≥70%,比表面积 1 000~1 200 m2/g,强度≥95%,宁夏平罗国宁活性炭有限公司;氯化汞(HgCl2)纯度99.9%,贵州大龙银星汞业有限责任公司;氯化钾(KCl,A.R),天津市科密欧化学试剂有限公司;盐酸(分析纯),重庆川江化学试剂厂。
1.2 超低固汞催化剂制备
超低固汞催化剂采用等体积浸渍法制备,按照实验需求分别把不同质量氯化汞和氯化汞-氯化钾,加入浓度为1 mol/L盐酸溶液中,在80℃浸渍6 h,采用阶梯升温干燥的方式,在80℃干燥2 h,90℃干燥2 h,100℃干燥2 h,120℃干燥至水分小于0.3%。
1.3 超低固汞催化剂的活性评价
超低固汞催化剂活性采用GC-9890B气相色谱仪进行评价,催化转化固定床模拟工业化装置长度为3 300 mm,内置Ø 45×3列管1根,触媒填装量约为2.3 kg,反应前通入氯化氢气体活化1 h,控制乙炔空间流速为30 h-1,乙炔与氯化氢流量采用流量计控制,流量比为1∶1.05,催化剂的活性用乙炔的转化率(xC2H2)作为评价指标,计算时把整个反应体系看作体积不变,总体积按照一个体积单位计算,计算式为:

式中:φAl为剩余乙炔的体积分数。
1.4 超低固汞催化剂的固汞效果评价
催化剂的固汞效果用氯化汞损失率[1]指标进行评价,加热催化剂,使其中的氯化汞升华,根据试样中氯化汞减少的质量占加热前试样中氯化汞质量的百分数计算氯化汞损失率。用四分法分取试样,称取约50 g,记为质量m0,精确至0.001 g,装入燃烧舟;在管式炉中通入氮气,控制流速约500 mL/min,将恒温炉升温至250℃并保持温度恒定,将装有试样的燃烧舟置于恒温管式炉中的恒温带区域恒温3 h,切断电源,继续通氮气,自然冷却至室温后,取出试样再次称其质量,记为m1,精确至0.001 g。氯化汞损失率计算式为:

式中:m0为试样的质量的数值,g;w0为试样中氯化汞的质量分数的数值,%;m1为250℃焙烧后试样的质量的数值,g;w1为250℃焙烧后试样中氯化汞的质量分数的数值,%。
2 使用性能机理分析
2.1 4%HgCl2/C体系催化机理

图1 4%HgCl2/C催化剂催化转化率
制备4%HgCl2/C超低固汞催化剂,催化性能见图1[2],在720 h内,其催化转化率为85%~86.5%,氯化汞损失率为6.3%,氯化汞为分子晶体,熔点276℃,其蒸汽压随温度变化见表1[3],在200℃下,氯化汞蒸汽压明显增大,因此,在250℃条件下测定氯化汞损失率时,其固汞效果较差。

表1 氯化汞蒸汽压与温度的关系
从汞的外层电子分析:见图2,汞原子的外层电子结构为3d106s26p0,当形成氯化汞后,失去6 s轨道上的2个电子,形成空轨道的6s06p0,当周围存在Cl-,在Cl-配位场的影响下而发生sp3杂化,具有4个sp3杂化轨道,形成四面体构型,图1(a)所示,4个Cl-和Hg2+以配位键的形式结合,便形成四面体构型的配位络合离子HgCl2-4,如图2(b)所示。
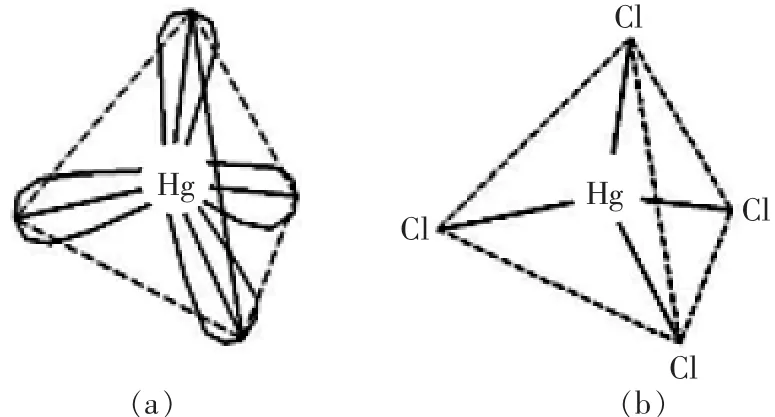
图2 sp3杂化轨道(a)和HgCl2-4络合结构(b)

表2 汞、钾原子半径及电子排布
乙炔的结构特点[3]为:2个三键碳均为sp杂化,每个碳还各剩2个互相垂直的p轨道,每个轨道上都有一个电子,2个三键碳原子各用1个sp杂化轨道经轴向重叠形成1个碳碳σ键,再各用2个p轨道经侧面形成两个碳碳π键。碳碳三键是由1个σ键和2个π键共同组成。由于π键是经侧面重叠形成的,不能重叠的很充分,所以π键的键能比σ键低,较容易打开。
当乙炔分子接近络合离子HgCl2-4时,电子从乙炔这边移向金属Hg2+的一边,围绕着2个C和1个Hg原子核运动,乙炔的π键发生断裂,由于同一个平面上的Hg2+有一个充满2个电子的dxx轨道,乙炔的反键π*轨道能级与几何构型接近,因而发生局部的重叠,形成了σ-π配键。其活性中心Hg2+一边从乙炔成键π轨道中拉走电子,一边又向乙炔的反键π*轨道“反馈”电子。这样一拉一推使乙炔与极性分子氯化氢的加成反应较为容易进行,其具体反应机理如下。
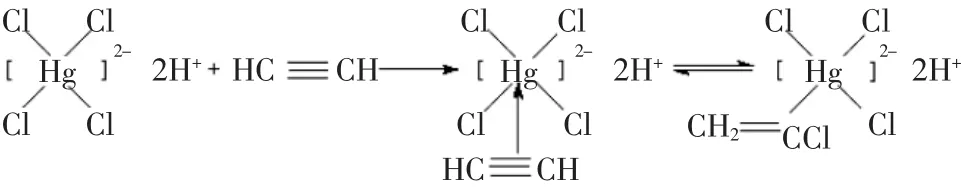
(2)汞元素作为给电子基团,与汞元素相连的双键碳原子电子云密度较大,H+优先与双键碳原子发生亲电加成反应,C-Hg键断裂,产生氯乙烯单体,其反应如下:
2.2 HgCl2-KCl/C体系催化机理
制备4%HgCl2-3%KCl/C超低固汞催化剂,测定其氯化汞损失率为2.1%,催化性能见图3。

图3 4%HgCl2-3%KCl//C催化剂催化转化率
图3所示,在720 h内转化率在89%~90%波动,与4%HgCl2/C相比,其催化转化率明显提高,其原因为:(1)单独负载氯化汞时,由于分子间作用力,氯化汞以分子团簇的形式负载于载体活性炭表面,活性组分分散度低,氯化汞损失率较高,固汞效果较差,当络合离子HgCl2-4与K+形成配合物K2HgCl4,以配合物K2HgCl4的形式负载于载体活性炭表面,减小了氯化汞分子间作用力,提高了分散度,催化转化率明显提高,抑制了氯化汞的流失,增强了固汞效果。(2)如表2所示,钾金属离子无d电子,具有较强的给电子能力,钾的加入使汞周围的电子云密度增大[5,6],有利于吸附反应物乙炔,提高了催化转化率。
同理,乙炔与氯化氢在HgCl2-KCl催化体系反应时,按照如下机理进行。
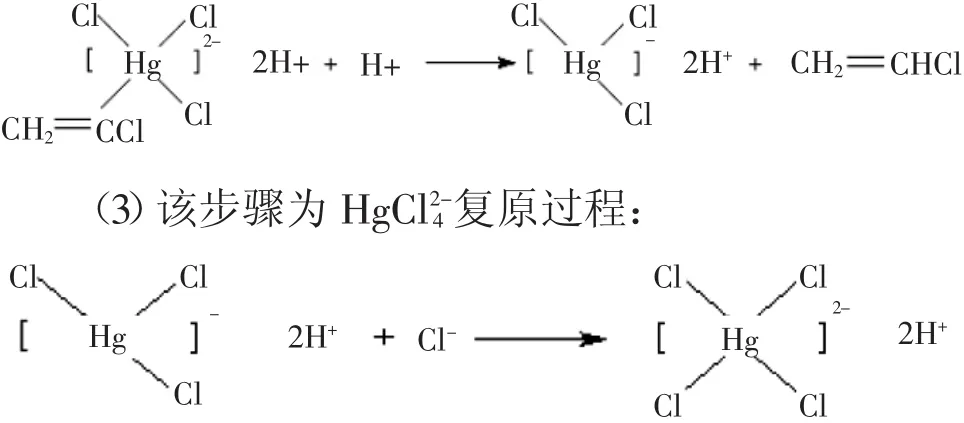
(1)乙炔进攻K2HgCl4,将π电子转移到活性中心汞元素上,乙炔中的π键断裂,形成C-Hg键,发生如下反应:

(2)汞元素作为给电子基团,与汞元素相连的双键碳原子电子云密度较大,H+优先与双键碳原子发生亲电加成反应,C-Hg键断裂,产生氯乙烯单体,其反应如下:

3 结论
以氯化汞为主催化成分,氯化钾为助剂,活性炭为载体制备了乙炔氢氯化超低固汞催化剂,氯化汞与氯化钾可形成配合物K2HgCl4,其催化机理为络合催化,氯化钾的加入有效增加了活性组分氯化汞的分散度,提高了乙炔氢氯化转化率,抑制了氯化汞流失,提高了使用性能。
[1]GB/T31530-2015,氯乙烯合成用低汞触媒.北京;李法曾,杨秀玲,那风环,等.2015.1-8.
[2]颜才南,胡志宏,曾建华.聚氯乙烯生产与操作.北京,化学工业出版社,2014,102-103.
[3]邢其毅,裴伟伟,徐秋瑞,等.基础有机化学.北京,高等教育出版社,2005,369-370.
[4]何正华.Au基催化剂催化乙炔氢氯化反应的机理研究.天津,天津大学,2012.
[5]王声洁,沈本贤,肖卫国.乙炔氢氯化高分散载金催化剂的制备及催化性能.石油学报(石油加工),2010 26(2);201-207.
[6]张善正.AuCl3及多种复合催化剂表面的乙炔氢氯化反应机理研究.新疆,石河子大学,2013.
Study on the mechanism of K on the preformance of ultra-low fixed mercuy catalytic system for hydrochlorination of acetylene
YU Yan-fang1,LI Wu-bin2,ZHANG Bin2
(1.Guizhou Province Real Estate Registration Center, Guiyang 550003,China;2.Guizhou Silver Star Group, Guiyang 550000,China)
The ultra-low fixed mercury HgCl2-KCl/C catalysts were prepared by the impregnation method,the catalytic performance was investigated in fixed bed reactor and fixed mercury effect was assessed in constant temperature furnace.The results show that KCl and HgCl2can form complexes K2HgCl4,the catalytic mechanism is complex catalysis,the dispersion of active was increased,catalytic performance was enhanced and fixed mercury was strengthened.
ultra-low fixed mercury catalysts;mechanism;complexes;complexe catalysis
TQ314.24+2
B
1009-1785(2017)12-0001-03
2017-09-18
