靶向ezrin增强子关键区的CRISPR/Cas9载体的构建
2017-12-19郭晓龙张青峰野庆松莫镇涛李文娜高书颖
郭晓龙, 张青峰, 野庆松, 莫镇涛, 李文娜, 高书颖
(遵义医学院珠海校区 生物工程系, 珠海 519041)
靶向ezrin增强子关键区的CRISPR/Cas9载体的构建
郭晓龙, 张青峰, 野庆松, 莫镇涛, 李文娜, 高书颖
(遵义医学院珠海校区 生物工程系, 珠海 519041)
构建靶向人ezrin增强子关键区的CRISPR/Cas9载体并检测其基因敲除功能。设计2个gRNA,分别靶向人ezrin增强子关键区的上、下游。合成gRNA寡核苷酸序列,连接至质粒pX459构建重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER。将鉴定正确的CRISPR/Cas9重组质粒共转染至食管癌EC109细胞中,提取细胞基因组DNA,针对gRNA靶位点两侧进行PCR扩增和亚克隆测序分析。经限制性内切酶酶切和测序鉴定表明,CRISPR/Cas9重组质粒构建正确。在共转染重组质粒的细胞基因组DNA中检测到ezrin增强子关键区的缺失。成功构建了靶向人ezrin增强子关键区的CRISPR/Cas9载体,能够实现目标序列的定向敲除。
靶向敲除;ezrin增强子;CRISPR/Cas9
CRISPR/Cas(clustered regularly interspaced short palindromic repeats-associated)是很多细菌和大部分古生菌的天然免疫系统,通过对入侵的病毒和核酸进行特异性的识别,利用Cas蛋白进行切割,从而达到对自身的免疫[1]。CRISPR/Cas9系统借鉴细菌的防御策略,由gRNA(guide RNA)寻找特定的DNA序列,然后利用Cas9核酸内切酶对靶DNA进行切割,造成双链断裂,在没有模板的情况下,发生非同源末端连接,造成DNA缺失突变[2]。目前,CRISPR/Cas技术已广泛应用于多种细胞和模式生物的基因编辑[3-5]。
肿瘤相关基因ezrin在人食管癌[6]、胰腺癌[7]、前列腺癌[8]、乳腺癌[9]等多种肿瘤中存在异常表达现象,其表达上调与肿瘤细胞的移动侵袭相关。以往采用双荧光素酶报告基因检测系统研究发现,在人ezrin编码区的上游存在启动子和增强子区,ezrin增强子关键区-1297/-1186序列很可能对于Ezrin在肿瘤细胞中的高表达起重要作用[10-11]。人ezrin增强子关键区为非转录区域,不能用传统的RNA干扰的方法进行研究。本研究拟采用CRISPR/Cas9系统,在人ezrin增强子关键区的上游和下游分别设计筛选1个特异性gRNA靶位点,构建靶向ezrin增强子关键区的CRISPR/Cas9载体,对2个靶位点同时进行双链断裂,实现人ezrin增强子关键区的靶向敲除,为进一步构建基因编辑细胞系,研究ezrin增强子的生物学功能奠定基础。
1 材料和方法
1.1 质粒、细胞及主要试剂
质粒pX459购自Addgene公司;食管癌EC109细胞购自中国科学院上海细胞库;感受态细菌DH5α由本实验室保存;质粒抽提及PCR纯化回收试剂盒购自Axygen公司;LipofectamineTM2000转染试剂购自Invitrogen公司;Guide-it mutation detection kit购自Clontech公司;BbsI核酸内切酶购自NEB公司;pMD18-T载体、AgeI核酸内切酶、T4 DNA连接酶、PCR Mix购自Takara公司;Annealing Buffer(10×)购自Origene公司;胎牛血清购自Hyclone公司;DMEM培养基和Opti-MEM培养基购自Gbico公司。引物合成和测序由Lifetech公司完成。
1.2 gRNA靶位点设计及寡核苷酸链的合成
从GenBank中查找人ezrin序列(http://www.ncbi.nlm.nih.gov/gene/7430),利用在线软件http://www.e-crisp.org/E-CRISP/设计gRNA靶位点,分别位于人ezrin增强子关键区(-1297/-1186)的上游和下游。gRNA对应的寡核苷酸链(Oligo DNA)按照5′-G(N)20NGG-3′的PAM结构(protospacer adjacent motif)为设计原则,选择分值较高的序列,如果序列的正向寡核苷酸链(Forward oligo)5′端第一个碱基不是G,则在5′端添加一个G,相应地在反向寡核苷酸链(Reverse oligo)的3′端添加一个C。同时在每对互补序列的正向寡核苷酸链的5′端添加CACC,反向寡核苷酸链的5′端添加AAAC,使其退火后形成的末端与pX459经BbsⅠ酶切后形成的黏性末端互补。本研究合成的gRNA对应的Oligo DNA见表1,gRNA-EL和gRNA-ER分别靶向ezrin的-1319/-1300和-1192/-1173序列,NGG分别位于-1319上游和-1173下游。
1.3 pX459-gRNA重组质粒构建
将Oligo DNA稀释至终浓度为100 μmol/L,进行退火反应。反应体系如下:两条互补Oligo DNA各0.5 μL,Annealing Buffer(10×)2 μL,ddH2O 17 μL。将以上体系瞬时离心后,置于65℃水浴中孵育10 min,随后取出,室温下缓慢冷却1~2 h。取2 μL杂交后的双链DNA与载体pX459的BbsⅠ酶切回收片段连接,连接产物转化大肠杆菌DH5α感受态细胞,在氨苄青霉素抗性平板上筛选阳性克隆,经PCR进一步筛选后,提取重组质粒进行酶切和测序鉴定。测序鉴定引物为pX459序列,5′-CCAAGTAGGAAAGTCCCATAAG-3′。

表1 gRNA对应的寡核苷酸序列Table 1 Oligo DNA for gRNA
ezrin序列用大写字母表示,在序列两端添加的碱基用小写字母表示
1.4 细胞培养及质粒转染
人食管癌EC109细胞在含10%灭活胎牛血清的DMEM培养基中贴壁生长,用含0.25%胰蛋白酶和0.02% EDTA的消化液消化细胞,进行传代培养。将细胞接种于96孔细胞培养板,当细胞汇合率为60%~80%时,进行质粒瞬时转染,转染步骤参照LipofectamineTM2000转染试剂说明进行。
1.5 细胞基因组DNA突变位点PCR测序鉴定
质粒转染EC109细胞48 h后收取细胞,用Guide-it mutation detection kit进行基因组DNA PCR扩增。PCR引物列位于拟删除序列的上、下游,引物序列如下,PCR-F:5′-CACAAACGTGCCACTTAACCA-3′(ezrin-1543/-1523序列);PCR-R:5′-AACCGTCAAGCCTTTGAGAAA-3′(ezrin-798/-778序列)。将PCR产物连接至pMD18-T载体,连接产物转化大肠杆菌DH5α感受态细胞,在氨苄青霉素抗性平板上筛选阳性克隆。随机取20个克隆进行测序鉴定。测序引物为M13F(-47):5′-CGCCAGGGTTTTCCCAGTCACGAC-3′。
2 结果
2.1 CRISPR/Cas9重组质粒的酶切鉴定
gRNA-EL和gRNA-ER分别识别ezrin增强子关键区(-1297/-1186)的上游(-1319/-1300)和下游序列(-1192/-1173),其对应的oligo DNA经退火反应后分别与质粒pX459的BbsⅠ酶切回收片段与连接,构建CRISPR/Cas9重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER。质粒pX459含1个AgeⅠ酶切位点,2个BbsⅠ酶切位点。重组质粒pX459-sgRNA-EL含1个AgeⅠ酶切位点,无BbsⅠ酶切位点;pX459-sgRNA-ER含2个AgeⅠ酶切位点,无BbsⅠ酶切位点。经AgeⅠ单酶切和经BbsⅠ/AgeⅠ双酶切后,pX459能够分别形成9175 bp 1条带和8185 bp+972 bp+18 bp 3条带片段;而pX459-sgRNA-EL则形成相同的9178 bp 1条带;pX459-sgRNA-ER形成相同的8183 bp+994 bp 2条带。重组质粒酶切鉴定结果(图1)显示,酶切片段位置与预计一致。

图1 重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER的酶切鉴定Fig 1 Identification of recombination plasmids pX459-sgRNA-EL and pX459-sgRNA-ER by restriction enzyme
M:DL 10000 DNA marker;1:pX459经AgeⅠ单酶切;2:pX459经BbsⅠ/AgeⅠ双酶切;3:pX459-sgRNA-EL经AgeⅠ单酶切;4:pX459-sgRNA-EL经BbsⅠ/AgeⅠ双酶切;5:pX459-sgRNA-ER经AgeⅠ单酶切;6:pX459-sgRNA-ER经BbsⅠ/AgeⅠ双酶切
2.2 CRISPR/Cas9重组质粒的测序鉴定
进一步对重组质粒进行测序分析,结果(图2)显示,分别位于人ezrin增强子关键区上游和下游的gRNA-EL和gRNA-ER模板序列在载体pX459上的连接位置和方向完全正确,重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER构建成功。

图2 重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER测序鉴定Fig 2 Sequencing analysis of recombination plasmids pX459-sgRNA-EL and pX459-sgRNA-ER
A:pX459-sgRNA-EL,实框为gRNA-EL模板序列;B:pX459-sgRNA-ER,虚框为gRNA-ER模板序列
2.3 重组质粒共转染EC109细胞
重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER共转染食管癌EC109细胞48 h后,不经抗性筛选直接收取细胞。以转染细胞基因组DNA为模板,PCR扩增包含ezrin增强子关键区在内的DNA序列。gRNA-EL和gRNA-ER识别序列分别位于ezrin增强子关键区的两侧,预计非突变基因组DNA扩增片段长766 bp(-1543/-778);突变基因组DNA删除147 bp(-1319/-1173),扩增片段长约619 bp。PCR产物琼脂糖凝胶电泳检测结果见图3。由于质粒转染细胞后未经药物抗性筛选,存在ezrin增强子关键区未被敲除的细胞,因此在对照组和转染组均能检测到与预计766 bp相符的目的条带,为ezrin增强子关键区非缺失突变片段。而转染组还检测到小于700 bp的微弱条带,有可能是CRISPR/Cas9系统在EC109细胞基因组上导致了定点突变,需要进一步鉴定。
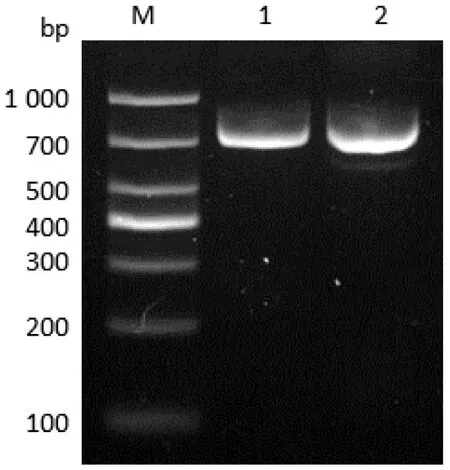
图3 转染细胞ezrin增强子关键区的PCR鉴定Fig 3 PCR amplification of ezrin enhancer key region from transfected cells
M:DL 1000 DNA marker;1:EC109细胞;2:共转染质粒pX459-sgRNA-EL和pX459-sgRNA-ER的EC109细胞
2.4 突变位点的测序鉴定
将共转染重组质粒的EC109细胞基因组PCR产物与pMD18-T连接,转化大肠杆菌DH5α感受态细胞,在氨苄青霉素抗性平板上筛选阳性克隆。随机取20个克隆进行测序鉴定。在测序的20个克隆中,有2个克隆(编号为CL-3和CL-20)在人ezrin增强子关键区发生了碱基的缺失。部分克隆的测序比对分析结果见图4,测序结果见图5,由图4和5中可见,CRISPR/Cas9重组质粒pX459-sgRNA-EL和pX459-sgRNA-ER分别在预定位点对基因组DNA进行的切割,实现人ezrin基因增强子关键区的靶向敲除。

图4 亚克隆测序的序列比对分析Fig 4 Alignment of subclones sequencing
阴影斜体为gRNA-EL靶位点,阴影正体为gRNA-ER靶位点
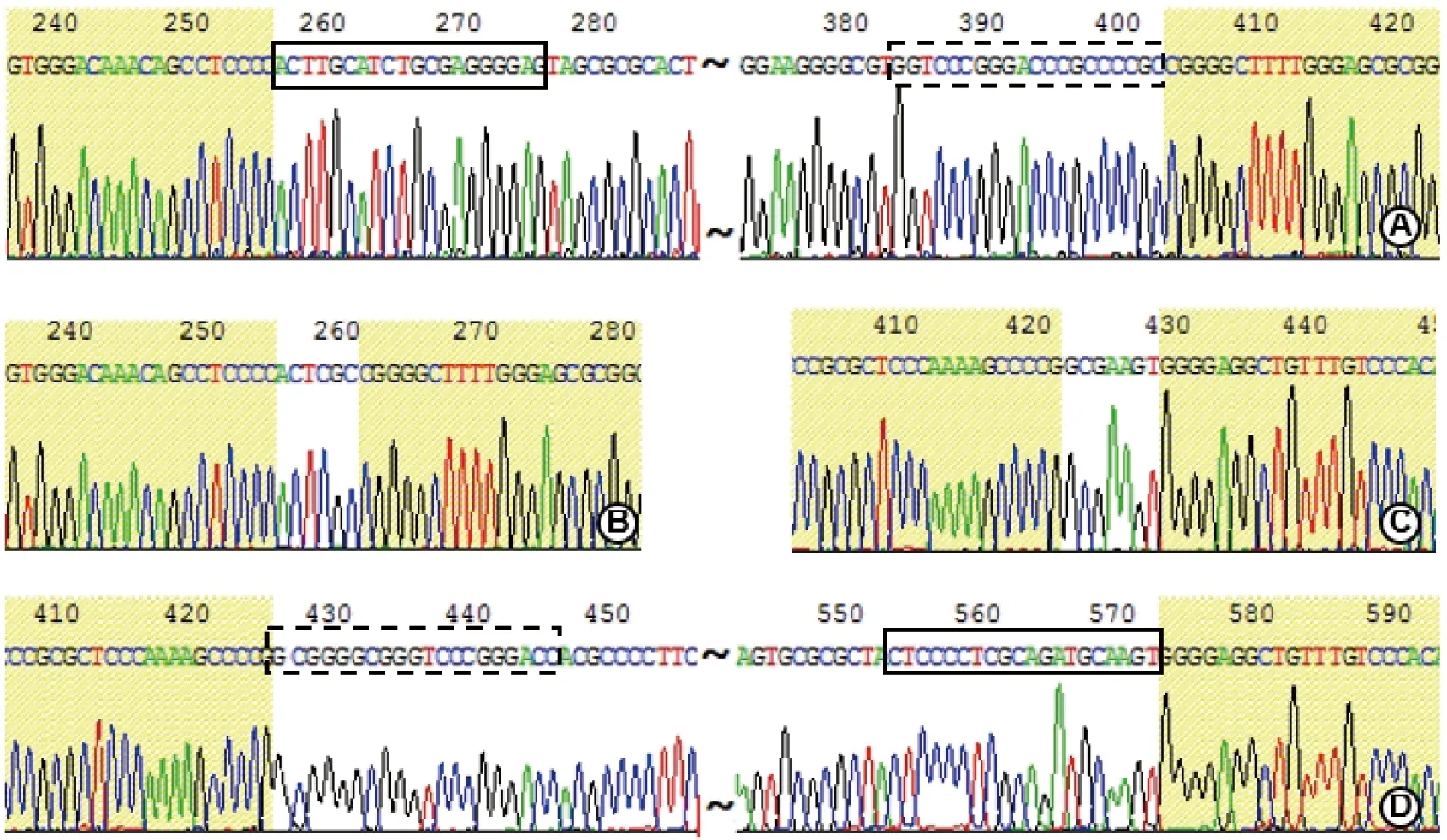
图5 亚克隆测序图Fig 5 The sequencing profile of subclones
A:克隆CL-10;B:克隆CL-3;C:克隆CL-20;D:克隆CL-19。黄色填充为ezrin增强子gRNA靶位点两侧序列,实框为gRNA-EL靶位点,虚框为gRNA-ER靶位点
3 讨论
对于基因增强子功能的研究通常采用报告基因的方法,将目标序列克隆至报告基因表达载体,根据报告基因的表达情况间接判断增强子功能。我们以往采用双荧光素酶报告基因检测系统研究肿瘤相关基因ezrin的表达调控机制,结果发现,位于基因启动子上游的-1541/-706序列具有转录增强作用[11-12],而-1297/-1186序列为增强子关键区[10,13],很可能对Ezrin在肿瘤细胞中的高表达起重要作用。采用报告基因方法时,仅克隆目标序列对其进行分离检测。实际上,细胞中增强子功能受其上下游序列影响,有可能会出现报告基因检测结果与实际情况不一致的现象。
随着近年来CRISPR/Cas9基因编辑技术的发展完善,由sgRNA引导Cas9核酸内切酶可以实现细胞中定向删除目标序列[2,14],使研究结果更为真实可靠。本研究构建的CRISPR/Cas9载体pX459-sgRNA-EL和pX459-sgRNA-ER,分别识别ezrin增强子关键区的上游和下游序列,靶向敲除DNA序列范围设计在增强子关键区的两侧。重组质粒转染食管癌细胞后,可以在预定范围敲除增强子关键区。后续工作将在本研究基础上,筛选靶向敲除ezrin增强子关键区的细胞系,研究增强子对Ezrin蛋白的表达及肿瘤细胞生物学行为的影响。
[1]CHYLINSKI K, MAKAROVA K S, CHARPENTIER E, et al. Classification and evolution of type II CRISPR-Cas systems[J]. Nucleic Acids Res, 2014, 42(10): 6091-6105.
[2]SHALEM O, SANJANA N E, HARTENIAN E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells[J]. Science, 2014, 343(6166): 84-87.
[3]CAO J, WU L, ZHANG S M, et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting[J]. Nucleic Acids Res, 2016, 44(19): e149.
[4]YE L, WANG J, TAN Y, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia[J]. PNAS, 2016, 113(38): 10661-10665.
[5]WALTON J, BLAGIH J, ENNIS D, et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma[J]. Cancer Res, 2016, 76(20): 6118-6129.
[6]XIE J J, XU L Y, WU Z Y, et al. Prognostic implication of ezrin expression in esophageal squamous cell carcinoma[J]. J Surg Oncol, 2011, 104(5): 538-543.
[7]ZHONG Z Q, SONG M M, HE Y, et al. Knockdown of ezrin by RNA interference reverses malignant behavior of human pancreatic cancer cells in vitro[J]. Asian Pac J Cancer Prev, 2012, 13(8): 3781-3789.
[8]阳 宁, 王 玲, 陈 仙, 等. 埃兹蛋白(ezrin)基因沉默抑制人前列腺癌PC-3细胞增殖和侵袭[J]. 细胞与分子免疫学杂志, 2016, 32(6): 821-824.
[9]HOSKIN V, SZETO A, GHAFFARI A, et al. Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion[J]. Mol Biol Cell, 2015, 26(19): 3464-3479.
[10]GAO S, DAI Y, YIN M, et al. Potential transcriptional regulatory regions exist upstream the human ezrin gene promoter in esophageal carcinoma cells[J]. Acta Biochim Biophys Sin, 2011, 43(6): 455-464.
[11]张青峰, 卫金岐, 张芳婷, 等. 几种肿瘤细胞中ezrin基因增强子区转录调控特性的研究[J]. 中国细胞生物学学报, 2014, 36(5): 610-616.
[12]GAO S Y, LI E M, CUI L, et al. Sp1 and AP-1 regulate expression of the human gene VIL2 in esophageal carcinoma cells[J]. J Biol Chem, 2009, 284(12): 7995-8004.
[13]GAO S Y, DAI Y P, LONG X, et al. Identification of the VIL2 enhancer in human embryonic kidney cells[J]. Cell Biol Int, 2011, 35(10): 967-971.
[14]RAN F A, CONG L, YAN W X, et al. In vivo genome editing usingStaphylococcusaureusCas9[J]. Nature, 2015, 520(7546): 186-191.
ConstructionofCRISPR/Cas9vectorstargetingtoezrinenhancerkeyregion
GUO Xiao-long, ZHANG Qing-feng, YE Qing-song, MO Zhen-tao, LI Wen-na, GAO Shu-ying
(Department of Bioengineering, Zhuhai Campus of Zunyi Medical University, Zhuhai 519041, China)
The aim of this study is to construct CRISPR/Cas9 vectors, which targets the key region of human ezrin enhancer, and assays knockout function of the vectors. Two gRNAs, aiming at upstream and downstream of human ezrin enhancer key region respectively, were designed, synthesized and ligated into plasmid pX459 to construct pX459-sgRNA-EL and pX459-sgRNA-ER. Then, the correct CRISPR/Cas9 recombinant plasmids were co-transfected into human esophageal carcinoma EC109 cells. Genomic DNA was extracted for PCR amplification of a region flanking the CRISPR target sites and subclonal sequencing analysis. By restriction enzyme digestion and DNA sequencing assay, it was confirmed that CRISPR/Cas9 recombinant plasmids were constructed successfully. Deletion of the ezrin enhancer key region was found in co-transfection cells genomic DNA. Therefore, CRISPR/Cas9 vectors targeting human gene enhancer key region were constructed and could create fixed sequence knockout.
target knockout; ezrin enhancer; CRISPR/Cas9
2016-10-31;接受日期2016-11-10
国家自然科学基金(31360212);贵州省科技厅联合基金(LKZ[2013]04);贵州省科学技术基金(黔科合J字[2014]2180号)
郭晓龙,专业方向为基因表达调控机制,E-mail: 1406049005@qq.com
高书颖,博士,教授,研究方向基因表达调控机制,E-mail:shuyinggao@163.com
10.3969/j.issn.2095-1736.2017.06.019
Q78
A
2095-1736(2017)06-0019-04
