Advances of Genetic Map Construction and QTL Mapping in Peanut
2017-12-19AddisuGETAHUNYANGXinleiHEMeijingCUIShunliMUGuojunLIULifeng
Addisu GETAHUN, YANG Xin-lei, HE Mei-jing, CUI Shun-li, MU Guo-jun, LIU Li-feng*
(1. North China Key Lab. for Crop Germplasm Resources of Education Ministry/Lab. for CropGermplasm Resources of Hebei, College of Agronomy, Agr. Uni.of Hebei, Baoding 071001, China;2. Ethiopian Institute of Agricultural Research, Pawe Agricultural Research Center, ET 25)
AdvancesofGeneticMapConstructionandQTLMappinginPeanut
Addisu GETAHUN1, 2, YANG Xin-lei1, HE Mei-jing1, CUI Shun-li1, MU Guo-jun1, LIU Li-feng1*
(1.NorthChinaKeyLab.forCropGermplasmResourcesofEducationMinistry/Lab.forCropGermplasmResourcesofHebei,CollegeofAgronomy,Agr.Uni.ofHebei,Baoding071001,China;2.EthiopianInstituteofAgriculturalResearch,PaweAgriculturalResearchCenter,ET25)
The construction of genetic maps and QTL mapping are of great significance for peanut molecular breeding. With the development of various molecular markers and sequencing technology, great advances have been made in genetic study for important quantitative traits in peanut. Some molecular markers associated with traits of disease-resistant and yield was developed. In this paper, the development of molecular markers, the construction of molecular genetic linkage maps, and QTLs mapping in peanut were reviewed, which provided theoretical references for the application of molecular marker assisted selection, the improvement of breeding efficiency, and the acceleration of breeding process in peanut.
peanut; genetic map; QTL mapping; molecular markers
1 Introduction
Cultivated peanut or groundnut (ArachishypogaeaL.), an allotetraploid (2n=4x=40) crop, is grown extensively nearly about 108 nations. More than two-thirds of peanut global production comes from seasonally rain field regions. Annual global area coverage was about 25.44 million ha with the total production of 45.22 million tons and average productivity was 1.676 t/ha. Asian and African countries were the leading producers with the production of 11.6 million ha per annum that accounted (47.15%) and 11.7 million ha per annum (47.56%) respectively holds maximum global area coverage. The productivity of Asia (2.217 t/ha) and Africa (0.929 t/ha) remains still very poor as compared to America (3.632 t/ha)[1].
Peanut is also valuable and very important crop for food security, oilseed and to generate cash. This versatile and precious crop is consumed as extracting cooking edible oil, fresh level consumption as a spike started from at farm gate level, boiled, roasted, herbal tea, peanut milk, used as raw material for different confectionary preparations, and peanut butter by human while fresh protein-rich fodder and haulm by livestock. In addition, it also plays a vital role in the improvement of soil fertility, soil productivity and making keep soil healthy through fixing atmospheric nitrogen. Economically important peanut cultivars possessing genetic resilience for abiotic and biotic stress with enhanced oil/haulm quality and pod yields required for increasing productivity.
Cultivated peanut has been faced an intensive artificial evaluation and selection process through conventional breeding systems resulting in undesirable changes in yield, disease resistance and other important traits[2-3]. Immensely and tremendous progress have been made in peanut genetic improvement for economic yield and yield component traits in the past few years[2]. The previous studies had been giving more attention on morphological and some selected important agronomical traits[4], but the integration of genomic tools, such as molecular markers with conventional breeding approaches has been a promise to overcome the genetic bottlenecks/challenges and enhance our breeding efficiency leading to the rapid development of improved cultivars with good consumption quality and desirable traits in modern molecular breeding scenarios. The availability of many thousands of molecular markers as simple sequence repeats (SSRs) in cultivated peanut are very few and informative. Good quality single nucleotide polymorphism (SNPs) molecular markers are available in cultivated peanut[5], but SNPs molecular markers have been particularly used to interpret polyploidy genotyping data than phenotypic. Other molecular technologies/tools like diversity array technology (DArTs) seem to be unveiling to the best for high throughput markers among other molecular technologies in peanut molecular breeding. DArT also provided genome-wide profiling of genotypic data at a lower cost and promptly to conduct comprehensive marker-trait association (MTA) analysis of many traits of interests[5-7]. Deployed of genomics-assisted breeding programs or marker assisted selection such as family-based genetic linkage mapping efforts were used to identify few quantitative trait loci (QTLs) particularly simply inherited traits with major phenotypic effect with QTLs for complex traits with low phenotypic effects[8]. Family-pedigree based trait mapping approaches has several limitations such as an inability to address multiple agronomic traits in the single population and also used very low-density genetic linkage maps and low QTL resolution power and also an overestimation of the phenotypic effect of QTLs[9]. Many economically important traits are quantitative in nature and have high complex behavior. Association studies with the integration of genome-wide markers would help us to construct high-resolution genetic linkage maps and be used to identify complex traits[7,10]. The objectives of this review paper: ① to evaluate, identify and assess the previously constructed genetic linkage maps and ② to evaluate OTLs and QTL mapping of peanut.
2 Development of molecular markers
DNA molecular markers were discovered in peanut molecular breeding and genetics for improvement and selection of better cultivars at a molecular DNA level. The most important points are screening of hundreds of lines in a breeding population simultaneously with the combination of thousands of different molecular markers. For instance, a device, called a chip, was developed and holds 60 000 DNA fragments in each individual wells (Unpublished peanut foundation report July 2015). DNA fragments from breeding lines that stick to in each wells and it can be traced to a point on the cultivated peanut genome maps. Phenotyping agronomic traits clearly showed that the positions of that given map were being an important pinpoint to achieving the breeding objectives. Molecular DNA marker assisted method can detect traits of polymorphism at a given locus between two or more parents in a genetic cross. The sequence of differences at the marker site is detected as differences in the length of fragments created by PCR or restriction incision enzymes. Among all genomics resources, molecular DNA markers had been used directly to construct genetic linkage maps and QTLs mapping in molecular level.
Now in this modern scientific world several molecular markers have been developed and available in cultivated peanut, for instance restriction fragment length polymorphisms (RFLPs), random amplified polymorphic DNAs (RAPDs), amplified fragment length polymorphisms (AFLPs) and diversity arrays technology (DArT). These molecular markers have been proved their utility from time to time[11]. SSR markers are multi-allelic, co-dominant and easy to be used than other mentioned molecular markers, but the SNP markers are highly amenable to high-throughput genotyping systems than SSR.
The development and application of SNP molecular markers are still not routine in crop cultivar development, especially not in low-tech laboratories due to the reason of some incompatibility and polymorphism restriction nature. DArT which has been developed at DArT Pty Ltd (Australia) in collaboration with ICRISAT (India), CIRAD (France), Catholic University of Brasília and EMBRAPA (Brazil) is very useful and powerful genomic tools of the molecular breeding program. DArT with a range of genotypes representing diploid (AA,BB) and tetraploid (AABB) genome species showed a very low level of polymorphism in tetraploid genotypes and moderate levels of diversity among accessions from diploid genome species[6,12]. A few research reports indicated that DArT molecular marker technology may not be applicable for breeding programs in cultivated peanut because of its low polymorphic nature, but it is very important for mastering of the genome transferring of important traits from wild relatives into a cultivated peanut.
Some research results revealed that a total of 3 371 SSR markers from different resources of both published and unpublished were used to screen the polymorphism between two parental genotypes and polymorphic SSRs which were used to genotype the F2population of peanut[13]. But now the number of tested SSR molecular markers was being increased and have more than 9 274[14-15]. Those SSR molecular markers with the prefixes of pPGPseq, pPGSseq, TC, IPAHM, Ah, RI, EE, EM, GA, GM, GNB, AC, Ad, ARS, gi, AHBGS, PM, AHS, AHGS and HAS have been developed previously by different scientists and still being used to test in many research fields[16-29]. SSR molecular markers with prefixes of XY and POCR were developed and published by Tang and his colleague's laboratory[30], and those with prefixes AGGS were developed by Huang and his coworkers' laboratory[13]. Based on the origin and nature of molecular DNA marker applications SSR markers were classified into four categories: genomic SSRs (1 467), EST-SSRs (1 589), BAC-end SSRs (155) and transcript-SSRs (160)[13].
3 Study on genetic linkage maps
3.1 Mapping populations
In peanut, genetic population mapping systems had been conducted by a different scientist at different times via the use of different mechanisms depending on their research objectives and by looking the existed research gaps and the availability of research materials. The previous research reports stated that three recombinant inbred line (RIL) populations, RIL-1 (TAG 249 ICGV 86031), RIL-2 (ICGS 769 CSMG 84-1) and RIL-3 (ICGS 449 ICGS 76) were developed and genotyping by ICRISAT, in India and the genetic linkage map had been constructed between those inbreed line populations[31]. However, the RIL-1 populations developed previously before developing the rest two inbreed lines and QTL mapping of those inbred lines had been reported by ICRISAT scientists[12, 32]. Populations of RIL-2 and RIL-3 totally consisted of 177 and 188 individual RIL populations respectively. RIL-2 and RIL-3 had been investigated for the purpose of developing high-density genetic linkage maps and QTL mapping analyses. In this research program, scientists identified drought tolerant QTLs traits in normal water and under stress conditions, and 153 main QTLs (M-QTL) and 25 epistatic/interaction QTLs (E-QTL) traits had been identified for drought resistant and they were genetically mapped in 20 linkage groups in this research. 82 RILs derived from the cross between peanut genotypes Yuanza9102 and Chico were employed as genetic linkage mapping population by Pengetal[33]. Zhang developed a RIL derived from a cross of Zheng8903 and Yuhua4 and invested 20 traits related to yield, quality and disease[34]. In addition to the above mapping populations other scientists like Bertioli and his collaborators developed recombinant inbred line populations from distinctive F1generations by using genetically cloned plants and finally they had isolated planting materials by cutting to produce enough F2seeds and selected single-seed descent breeding methods to develop the F5/F6generations[35]. The F1generations already derived from the diploid populations which had been the cross betweenA.duranensisK7988 andA.stenospermaV10309[24, 36]. A genetic map consisting of 20 linkage groups was constructed by using the F2population derived from the cross between Fuchuan Dahuasheng and ICG637[37]. Lietalused F5/F6generations as mapping population to construct a genetic linkage map[38]. Other scientists also developed mapping populations by using backcross with filial (f) generations and RIL with backcross populations or breeding lines.
3.2 Genetic linkage map construction
The challenges of peanut molecular genetics and breeding have been reported that a very low polymorphism nature, so the generation of informative genetic markers has been very difficult to apply on peanut and this has been a fundamental limitation of peanut improvement at molecular DNA level. The first genetic map of peanut was constructed based on phenotypic traits, but modern genetic maps are being constructed based on DNA molecular levels by using genotype data.
Genetic map constructions of the cultivated peanut genomes are needed to show the locations and positions of all genes and to discover useful DNA molecular markers for those genes identification from their specific target locus. As part of the genome maps, assembled and connected sequences may be visualized as a series of small ''Gene Islands''. A new technology called 'Moleculo' was reported and used as a bridge between the small islands, then creating larger islands and finally, it can be joined into a continent. A complete cultivated peanut genome maps will be constructed by this way (unpublished Peanut foundation report July 2015). However, assembling more detailed maps of the cultivated peanut genome especially focused on the ability to distinguish the pieces of DNA that came from each of the two wild parent genomes.
Construction of a high-quality and dense genetic linkage maps, applying one or more molecular markers, can be detected high levels of polymorphism between individual populations and their respective parents. The first molecular map of peanut was constructed between the diploidsA.stenospermaKrapov. andW.C.GregoryxandA.cardenasiiKrapov. andW.C.Gregory[39]by the application of Restriction Fragment Length Polymorphism markers (RFLPs) and to associate 117 markers into 11 linkage groups. Additional maps were also subsequently published by applied Randomly Amplified Polymorphic DNA (RAPD)[40]and Simple Sequence Repeats (SSRs) molecular markers[24-25]. The first tetraploid map in peanut had been constructed based on the development of 370 RFLP loci across 23 linkage groups in the complex interspecific cross between Florunner × 4x [A.batizocoiKrapov. andW.C.Gregory(A.cardenasii×A.diogoiHoehne)]. Another inter-specific tetraploid linkage map of peanut also constructed by the investigation of 298 loci and 21 linkage groups which were derived from a backcross betweenA.hypogaeaand a synthetic amphidiploid[41]. And also in peanut another RFLP-based genetic linkage map was published by used BC1 tetraploid population which was derived from a synthetic allotetraploid [A.batizocoi× (A.cardenasii×A.diogoi)]4xcrossed with cultivated peanut[42]. This tetraploid map was particularly informative to genome structure because it was allowed the assignment of marker alleles on A or B genomes by reference to the known genomes of the diploid parents of the synthetic allotetraploid crop species. These above-mentioned research reports revealed that marker order was highly conserved between the A and B genomes. Recently the linkage maps also constructed from the crosses betweenA.hypogaeagenotypes. Wangetal[43]constructed a genetic linkage map of cultivated peanut which was 2 119.4 cM in length with an average distance of 9.55 cM and contained 223 loci located in 22 linkage groups. Based on the bacterial wilt resistance evaluation of F6and F7RILs and DNA marker, Jiangetal[44]developed a map including 8 linkage groups covering a map distance of 603.9 cM. Hongetal[45]developed a genetic linkage map consisting of 108 SSR loci (102 genomic-SSR and 6 EST-SSR) in 20 linkage groups using 184 recombinant inbred lines derived from a cross between two Spanish type peanut cultivars (Yueyou13 × Fu95-5) as mapping population. Exceptional cases that the most recently published genetic linkage map containing 1 114 loci across traits and 21 linkage groups can construct highly polymorphic markers derived from sequences harboring miniature by the inverted of repeat transposable elements[27]. Therefore, there is a continuing need to generate high dense genetic linkage maps for the cultivated tetraploid peanut that will not only cluster the markers into the expected 20 linkage groups to cover the haplotype chromosome numbers, but it also facilitated marker-trait association and eventually to assist genetic improvement research progress in allotetraploid peanut.
The first microsatellites map in peanut was applied from a cross betweenA.duranensisandA.stenosperma[24]. This genetic map had comprised 11 linkage groups covering 1 230 cM. Subsequently, a microsatellite map of the B genome based on a cross ofA.ipaensisand the closely relatedA.magnamap also had 10 linkage groups with 149 loci spanning length and the total map distance of 1 294 cM. Bertioli and his colleagues research report stated that the comparison of 51 shared markers between these two maps which was mentioned in the above revealed that high levels of synteny with all linkage groups, but one of the B linkage groups showed a single main correspondence to A linkage group; this seemed largely consistent with the observations for the previously mentioned tetraploid molecular maps of cultivated peanut. The main differences observed in the tetraploid study was one large B linkage group showed no marker correspondences to the A genome. The most recently published version had told us that 369 molecular markers in the 10 linkage groups were developed and the total genetic distance covered by this map was more than 3 000 cM[35]. Considering comparisons with subsequently published linkage maps[40], it is possible to conclude that this distance was overestimated of several folds probably due to the mixing of different dominant and co-dominant marker types simultaneously (Bertiolietal., 2012 unpublished press overview of peanut genome structure).
Through large-scale screening of simple sequence repeat (SSRs) molecular markers, a sufficient number of polymorphic markers were identified for the generation of the first genetic linkage map based on cultivated x cultivated crosses[12, 26]. These maps were very useful for advanced breeding programs because they were incorporate and integrate QTLs for economically important traits such as disease resistance and drought-related traits. The result of Nagy (A high-density genetic linkage maps ofA.duranensis) and his collaborators indicated that a high-density genetic map had been constructed by using the program of MST Map[46]which were used to build a core genetic maps including all co-dominant markers using the cut-off p-value of 10~12 for clustering markers into linkage groups of families and their offspring or family-pedigree based mapping[47]. This above explained genetic map was constructed by the development of 1 673 co-dominant markers and other 1 723 different molecular markers.
3.3 Consensus genetic map construction
A consensus genetic map was constructed through the integrating of all component genetic maps using common molecular markers across different genetic linkage maps by using Merge Map computer program. Grouping of different LGs from component genetic maps used for construct consensus genetic maps. While integrating of component genetic maps, some differences have been observed in the names and nature of markers available on more than one mapping populations. Based on the common marker application systems and the comparison between component genetic maps, most of the linkage groups are consistent among the individual maps with a few exceptions, basically, these resources are accessed in http://cmap.icrisat.ac.in/cmap/sm/gn/gautami/databases. In the ICRISAT, India database a total of 542 markers are unique molecular markers i.e. applied to construct consensus genetic maps and it mapped only in one mapping population, while the remaining 355 markers were common, i.e. they were mapped in at least two mapping populations for instances187 markers were common between two maps, 72 markers were between three maps, 57 markers were between 4 maps, 20 markers were between 5 maps, 13 markers were between 6 maps, 3 markers were between 7 maps, 2 markers were between 8 maps and the remaining 1 molecular marker was common between 9 genetic maps and it has also served as anchor points for map integration and development. Therefore, a total of 355(39.58%) molecular markers are anchor markers which presented in all 20 LGs in this consensus genetic map. The remaining 542(60.42%) markers were unique for each individual mapping population (http://cmap.icrisat.ac.in).
4 QTL mapping
Agronomic and yield-related traits are almost always quantitative traits in plant species. QTL mapping has been widely undertaken in various crops to detect the genomic regions controlling important traits. Because of a large genome and low genetic diversity, the progress in QTL mapping is hindered in peanut. However, along with the rapid development of molecular marker technology, many studies have been conducted to identify QTLs for a wide range of various traits in the molecular mapping of the peanut in recent years. Selvarajetal[48]identified 5 SSR markers associated with pod and kernel traits in cultivated peanut using a bulked segregant analysis. This is the first report on the identification of SSR markers linked to pod- and kernel-related traits in cultivated peanut. Liuetal[49]reported 4 and 6 QTLs controlling main stem height and 5 and 8 QTLs controlling branch length were detected based on
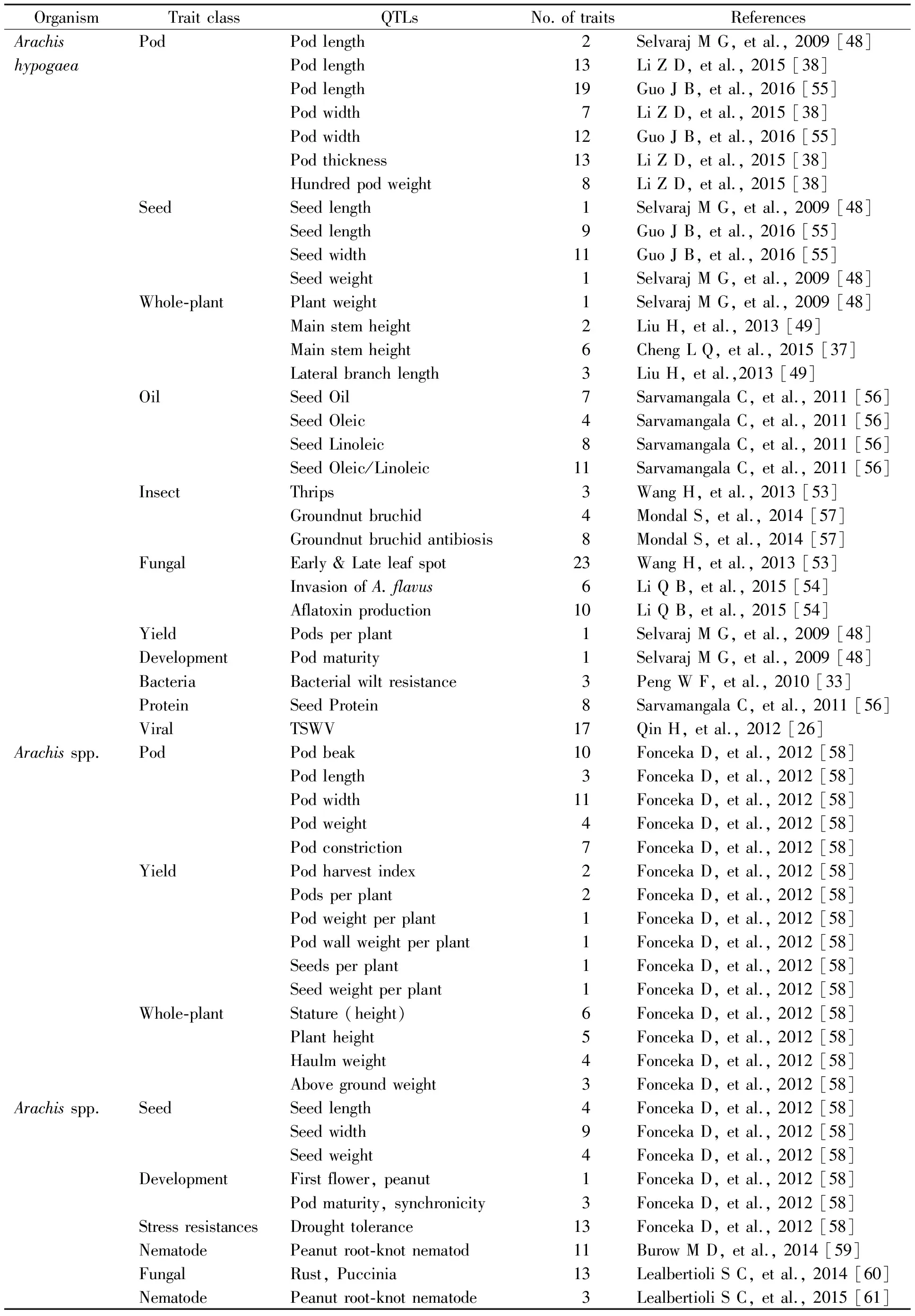
Table 1 Identified QTLs and trait class in peanut
(Source: IPGI (http://peanutbase.org/files/genomes/, USA, University of Georgia, 2016)
In peanut database still, 11 QTLs databases were set up by international peanut genome institute in UGA, USA, and the total genome size was 491.38 kb.
the phenotypic data collected in Sanya and Yuanyang respectively. Pandey and his fellow's research report stated that 300 peanut germplasms were collected from 48 nations in worldwide across different agro-ecologies and those collected genetic resources were planted on different research fields with the included of 4 known checks[7]. From the above explained comprehensive research experiment, the researchers evaluated and identified economically important quantitative traits and they reported many QTLs from 524 marker-trait associations in 50 trait class especially 36 were the most economically important traits from different research environments have been isolated and mapped. Lietal[38]detected a total of 41 QTLs in two environments, including 13 for pod length, 7 for pod width, and 13 for pod thickness and 8 for hundred pod weight and a total of 6 QTLs in both environments, which including three for pod 1 for pod thickness and 2 for hundred pod weight. Chenetal[50]conducted QTL analysis by using two F2:3populations derived from cultivar Fuchuan Dahuasheng×ICG 6375 (FI population) and cultivar Xuhua13×cultivar Zhonghua6 (XZ population) and 39 QTLs were detected in two populations. Novel QTLs were identified, which related to pod length, pod width, seed length and width. Zhang[34]mapped 62 additive QTLs related to shape, yield, quality (fat, protein, fatty acids) and the disease resistance to web blotch in peanut based on a RILs population derived from Zheng8903×Yuhua4. Many QTLs for late leaf spot (LLs), rust, thrips, tomato spotted wilt virus (TSWV) and QTLs for resistance to seed invasion ofA.flavusand for resistance to aflatoxin production were detected[51-54]. A study for the identification of candidate QTLs for drought tolerance related traits, Ravietal[32]identified 53 main-effect and 8 epistatic QTLs using QTL Cartographer and QTLNetwork, which provided a basis for drought tolerance breeding in groundnut.
In many research experiment conclusions indicated that the F2:3populations were an ideal population for mapping genotypes which can be used to designate QTL genetic effects as either additive or dominant effects on their off spring[13]. Many scientists research suggested most of the QTLs for agronomic and quality traits were determined by the dominance or else over dominance effects. Combined with the result of bacterial wilt resistance (BWr) evaluated, Pengetal[33]detected 3 QTLs (qBWr1, qBWr2, qBWr3) for peanut BWr. These 3 QTLs formed 2 QTL combinations (qBWr1/qBWr3 and qBWr2/qBWr3) with additive by additive epistasis effects which contributed to qBWr with 12.81% and 16.56% respectively. According to Huang and her colleagues effort, out of 36 QTLs for agronomic, biotic/abiotic resistance and quality related traits and four QTLs (11.1%) were exhibited no apparent dominance effects over the additive effects, seven QTLs (19.4%) had the mid-parent dominance effect and twenty-five QTLs (69.4%) had dominance degree effects (d) that was more than one QTL which were exhibiting over-parent heterosis for these QTLs[13].
Many research experiments agreed on the main objectives of molecular breeding was to identify marker-trait associations (MTAs) for agronomic, quality, biotic or abiotic resistance traits in their complex nature using a diverse panel of genotypes. 300 peanut genetic resources which were collected from 48 countries by Pandey and his co-friends revealed that a total of 134 marker-trait associations (MTAs) were identified with 20% PV for 15 traits across from the whole germplasms. A total of 30 significant allele effects for these 15 traits were identified and associated with 24 markers which they showed significant impact on these traits while the rest 9 markers were found to be associated with multiple traits[7].
The peanut genome had been successfully sequenced in international peanut genome institute (IPGI) in the USA by convened a group of international peanut geneticists from America, China, Brazil, India and Israel in Georgia university (http://peanutbase.org/files/genomes/) and this organized team conducted the whole peanut decoding and encoding sequences at the first time. More and more biological database resource of peanut will make it possible for the construction of saturated linkage maps and the fine mapping of QTL. In general, from the past efforts up to now, many QTLs were identified and sequenced. The following table comprises lists of sequenced peanut QTLs and traits of interest (Table 1 and Table 2).
[1] FAO (2015) FAO statistical database. http://fastat.fao.org/
[2] Yu S L. Peanut varieties and their pedigree in China [M]. Shanghai: Shanghai Science and Technology Press, 2010.
[3] Yu S L. Peanut genetics and breeding in China [M]. Shanghai: Shanghai Scientific and Technology Press, 2011.
[4] Ren X P, Jiang H F, Yan Z Y, et al. Genetic diversity and population structure of the major peanut (ArachishypogaeaL.) cultivars grown in China by SSR markers[J]. PLoS One, 2014, 9(2): 1-10.
[5] Varshney R K, Graner A, Sorrells M E. Genomics-assisted breeding for crop improvement [J]. Trends Plant Sci, 2005, 10: 621-630.
[6] Kilian A. DArT-based whole genome profiling and novel information technologies in the support system of modern breeding of groundnut// 3rdInternational Conference for Peanut Genomics and Biotechnology on ''Advances in Arachis through Genomics and Biotechnology (AAGB), 4-8thNovember 2008, Hyderabad, India.
[7] Pandey M K, Upadhyaya H D, Rathore A, et al. Genomewide association studies for 50 agronomic traits in peanut using the "reference set" comprising 300 genotypes from 48 countries of the semi-arid tropics of the world[J]. PLoS One, 2014, 9(8).
[8] Varshney R K, Mohan S M, Gaur P M, et al. Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics [J]. Biotech Adv, 2013, 31: 1120-1134.
[9] Gupta P K, Rustgi S, Kulwal P L. Linkage disequilibrium and association studies in higher plants: present status and future prospects [J]. Plant Mol Biol, 2005, 57:461-485.
[10] Varshney R K, Ribaut J M, Buckler E D, et al. Can genomics boost productivity of orphan crops? [J]. Nat Biotech, 2012, 30: 1172-1176.
[11] Gupta P K, Kumar J, Mir R R, et al. Marker-assisted selection as a component of conventional plant breeding [J]. Plant Breed Rev, 2010, 33:145-217.
[12] Varshney R K, Bertioli D J, Moretzsohn M C, et al. The first SSR based genetic linkage map for cultivated groundnut (A.hypogaeaL.)[J]. Theor Appl Genet, 2009, 118:729-739.
[13] Huang L, He H Y, Chen W G, et al. Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut (ArachishypogaeaL.)[J]. Theor Appl Genet, 2015, 128(6):1103-1115.
[14] Zhao Y L, Prakash C S and He G.Characterization and compilation of polymorphic simple sequence repeat (SSR) markers of peanut from public database[J]. BMC Res Notes, 2012, 5:362-369.
[15] Liu L, Dang P M and Chen C Y. Development and utilization of InDel markers to identify peanut (Arachishypogaea) disease resistance [J]. Front Plant Sci, 2015, 6: 988.
[16] Bravo J P, Hoshino A A, Angelici C, et al. Transferability and use of microsatellite markers for the genetic analysis of the germplasm of someArachissection species of the genusArachis[J]. Genet Mol Biol, 2006, 29 (3):516-524.
[17] Cuc L M, Mace E S, Crouch J H, et al. Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (A.hypogaea)[J]. BMC Plant Biol, 2008, 8:55.
[18] Ferguson M E, Burow M D, Schulze S R, et al. Microsatellite identification and characterization in peanut (A.hypogaeaL.)[J]. Theor Appl Genet, 2004, 108:1064-1070.
[19] Gimenes M A, Hoshino A A, Barbosa A V G, et al. Characterization and transferability of microsatellite markers of the cultivated peanut (Arachishypogaea)[J]. BMC Plant Biol, 2007, 7:9.
[20] He G, Meng R, Newman M, et al. Microsatellites as DNA markers in cultivated peanut (ArachishypogaeaL.)[J]. BMC Plant Biol, 2003, 3:3.
[21] Hopkins M, Casa A, Wang T, et al. Discovery and characterization of polymorphic simple sequence repeats (SSRS) in peanut [J]. Crop Sci, 1999, 39:1243-1247.
[22] Liang X Q, Chen X P, Hong Y B, et al. 2009. Utility of EST-derived SSR in cultivated peanut (ArachishypogaeaL.) and Arachis wild species[J]. BMC Plant Biol, 2009, 9 (1):35.
[23] Macedo S E, Moretzsohn M C, Leal-Bertioli S C M, et al. Development and characterization of highly polymorphic long TC repeat microsatellite markers for genetic analysis of peanut[J]. BMC Res Notes, 2012, 5: 86.
[24] Moretzsohn M, Leoi L, Proite K, et al. A microsatellite-based, gene-rich linkage map for the AA genome ofArachis(Fabaceae)[J]. Theor Appl Genet, 2005, 111 (6):1060-1071.
[25] Moretzsohn M C, Barbosa A V G, Alves-Freitas D M T, et al. 2009. A linkage map for the B-genome ofArachis(Fabaceae) and its synteny to the A-genome [J]. BMC Plant Biol, 2009, 9:40.
[26] Qin H, Feng S, Chen C, et al. An integrated genetic linkage map of cultivated peanut (ArachishypogaeaL.) constructed from two RIL populations [J]. Theor Appl Genet, 2012, 124: 653-664.
[27] Shirasawa K, Koilkonda P, Aoki K, et al. In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut [J]. BMC Plant Biol, 2012, 12:80.
[28] Wang H, Penmetsa R V, Yuan M, et al. Development and characterization of BAC-end sequence derived SSRs, and their incorporation into a new higher density genetic map for cultivated peanut (ArachishypogaeaL.)[J]. BMC Plant Biol, 2012, 12:10.
[29] Zhang J, Liang S, Duan J, et al. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (ArachishypogaeaL.)[J]. BMC Genom, 2012, 13:90.
[30] Tang M, Chen Y N, Ren X P, et al. Genetic diversity ofArachisaccessionsby EST-SSR from cultivated peanut (ArachishypogaeaL.)[J]. Acta Agron Sin, 2012, 38:1221-1231.
[31] Gautami B, Pandey M K, Vadez V, et al. Quantitative trait locus analysis and construction of consensus genetic map for drought tolerance traits based on three recombinant inbred line populations in cultivated groundnut (ArachishypogaeaL.)[J]. Mol Breeding, 2012, 30(2): 757-772.
[32] Ravi K, Vadez V, Isobe S, et al. Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (ArachishypogaeaL.)[J]. Theor Appl Genet, 2011, 122:1119-1132.
[33] Peng W F, Jiang H F, Ren X P, et al. Construction of AFLP genetic linkage map and detection of QTLs for bacterial wilt resistance in peanut (ArachishypogaeaL.)[J]. Acta Agric Boreali Sin, 2010, 25(6): 81-86.
[34] Zhang X Y. Inheritance of main traits related to yield, quality and disease resistance and their QTLs mapping in peanut (ArachishypogaeaL.)[D]. Hangzhou: Zhejiang University.
[35] Bertioli D J, Ozias-Akins P, Chu Y, et al. The use of SNP markers for linkage mapping in diploid and tetraploid peanut[J]. G3 (Bethesda, Md.), 2013, 4: 89-96.
[36] Leal-Bertioli S C M, José A C F V, Alves-Freitas D M T, et al. Identification of candidate genome regions controlling disease resistance inArachis[J]. BMC Plant Biol, 2009, 9: 112.
[37] Cheng L Q, Tang M, Ren X P, et al. Construction of genetic map and QTL analysis for main stem height and total branch number in peanut (ArachishypogaeaL.)[J] Acta Agron Sin, 2015, 41(6): 979-987.
[38] Li Z D, Li X P, Huang L, et al. Mapping of QTLs for pod size related traits in cultivated peanut (ArachishypogaeaL.)[J]. Acta Agron Sin, 2015, 41(9): 1313-1323.
[39] Halward T, Stalker H T, Kochert G. Development of an RFLP linkage map in diploid peanut species [J]. Theor Appl Genet, 1993, 87:379-384.
[40] Garcia G M, Stalker H T, Schroeder E, et al. A RAPD-based linkage map of peanut based on a backcross population between the two diploid species ofArachisstenospermaandA.cardenasii[J] .Peanut Sci, 2005, 32:1-8.
[41] Foncéka D, Hodo-Abalo T, Rivallan R, et al. Genetic mapping of wild introgressions into cultivated peanut: a way toward enlarging the genetic basis of a recent allotetraploid [J]. BMC Plant Biol, 2009, 9:103.
[42] Burow M D, Simpson C E, Starr J L, et al. Transmission genetics of chromatin from a synthetic amphidiploid to cultivated peanut (ArachishypogaeaL.) broadening the gene pool of a monophyletic polyploid species[J]. Genet, 2001, 159 (2):823-837.
[43] Wang Q, Zhang X Y, Tang F S, et al. Constrction of genetic linkage map of peanut (ArachishypogaeaL.) based on SRAP makers[J]. Chin J Oil Crop Sci, 2010, 32(3): 374-378.
[44] Jiang H F, Chen B Y, Ren X P, et al. Identification of SSR markers linked to bacterial wilt resistance of peanut with RILs[J]. Chin J Oil Crop Sci, 2007, 29: 26-30.
[45] Hong Y B, Liang X Q, Chen X P, et al. Construction of genetic linkage map in peanut (ArachishypogaeaL.) cultivars[J]. Acta Agron Sin, 2009, 35: 395-402.
[46] Wu Y, Bhat P R, Close T J, et al. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph [J]. PLoS Genet, 2008, 4:10.
[47] Nagy E D, Guo Y F, Tang S X, et al. A high-density genetic map ofArachisduranensis, a diploid ancestor of cultivated peanut [J]. BMC Genomics, 2012, 13:469.
[48] Selvaraj M G, Narayana M, Schubert A M, et al. Identification of QTLs for pod and kernel traits in cultivated peanut by bulked segregant analysis [J]. Electron J Biotech, 2009, 12(2): 1-10.
[49] Liu H, Zhang X Y, Han S Y, et al. Inheritance analysis and QTL mapping of main stem height and lateral branch length in peanut (ArachishypogaeaL.)[J]. Chin J Oil Crop Sci, 2013, 35(5):508-514.
[50] Chen W C, Jiao Y Q, Cheng L Q, et al.Quantitative trait locus analysis for pod- and kernel-related traits in the cultivated peanut (ArachishypogaeaL.) [J]. BMC Genet, 2016, 17: 25 doi:10.1186/s12863-016-0337-x.
[51] Khedikar Y, Gowda M, Sarvamangala C, et al. A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut (ArachishypogaeaL.)[J]. Theor Appl Genet. 2010;121(5):971-84.
[52] Sujay V, Gowda M V C, Pandey M K, et al. Quantitative trait locus analysis and construction of consensus genetic map for foliar disease resistance based on two recombinant inbred line populations in cultivated groundnut (ArachishypogaeaL.)[J]. Mol Breeding, 2012, 30(2) :773-788.
[53] Wang H, Pandey M K, Qiao L, et al. Genetic mapping and quantitative trait loci analysis for disease resistance using F2and F5generation-based genetic maps derived from 'Tifrunner' × 'GT-C20' in peanut[J]. The Plant Genome, 2013, 6(3): E1-E10.
[54] Li Q B, Yang C Y, Huang L, et al. Genetic linkage map and QTL analysis of resistance toAspergillusflavusinArachishypogaeaL.[J]. Chin J Oil Crop Sci, 2015, 37(5): 596-604.
[55] Guo J B, Huang L, Cheng L Q, et al. An integrated genetic linkage map from three F2populations of cultivated peanut (ArachishypogaeaL.)[J]. Acta Agron Sin, 2016, 42(2): 159-169.
[56] Sarvamangala C, Gowda M V C, Varshney R K. Identification of quantitative trait loci for protein content, oil content and oil quality for groundnut (ArachishypogaeaL.)[J]. Field Crops Research, 2011, 122:1 49-59.
[57] Mondal S, Hadapad A B, Hande P A, et al. Identification of quantitative trait loci for bruchid (Caryedon serratus Olivier) resistance components in cultivated groundnut (ArachishypogaeaL.)[J]. Mol Breeding, 2014, 33(4): 961-973.
[58] Fonceka D, Tossim H A, Rivallan R, et al. Fostered and left behind alleles in peanut: interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding [J]. BMC Plant Biol, 2012, 12(1): 1-16.
[59] Burow M D, Starr J L, Park C H, et al. Introgression of homeologous quantitative trait loci (QTLs) for resistance to the root-knot nematode [Meloidogynearenaria(Neal) Chitwood] in an advanced backcross-QTL population of peanut (ArachishypogaeaL.)[J]. Mol Breeding, 2014, 34:2 393-406.
[60] Lealbertioli S C, Cavalcante U, Gouvea E G, et al. Identification of QTLs for rust resistance in the peanut wild speciesArachismagnaand the development of KASP markers for marker-assisted selection[J]. G3-Genes Genomes Genetics, 2014, 5(7):61-82.
[61] Lealbertioli S C, Moretzsohn M C, Roberts P A, et al. Genetic mapping of resistance toMeloidogynearenariainArachisstenosperma: a new source of nematode resistance for peanut[J]. G3-Genes Genomes Genetics, 2015, 6(2):377-390.
2016-10-24
国家现代农业产业技术体系建设专项(CARS-14);国家自然科学基金项目(31471523);农业部引进国际先进农业科学技术计划(948 计划)项目(2013-Z65);河北省科技支撑计划(16226301D);河北省高等学校科学技术研究重点项目(ZD2015056)
Addisu Getahun (1989-),男,埃塞俄比亚人,河北农业大学硕士研究生,主要从事花生遗传育种研究。
花生遗传图谱构建与QTL定位研究进展
Addisu Getahun1, 2,杨鑫雷1,何美敬1,崔顺立1,穆国俊1,刘立峰1*
(1. 华北作物种质资源教育部重点实验室/河北省作物种质资源实验室,河北 保定 071001;2. Ethiopian Institute of Agricultural Research, Pawe Agricultural Research Center, ET 25)
遗传图谱构建与QTL定位对花生分子育种具有十分重要的意义。随着分子标记与测序技术的快速发展,极大地促进了花生重要数量性状的研究进程,并获得了一些与抗病、产量等重要性状相关联的分子标记。本文对国内外花生的分子标记开发、遗传图谱构建以及QTL定位的研究进展进行了综述,为花生分子标记辅助选择,提高花生育种效率,加速育种进程提供了理论参考。
花生;遗传图谱;QTL;分子标记
S565.2; Q755-103
A
10.14001/j.issn.1002-4093.2017.02.001
*通讯作者:刘立峰,教授,主要从事花生遗传育种研究。Tel: 0312-7528136; E-mail: lifengliucau@126.com
