浅谈欧洲药品管理局补充监测药品制度
2017-12-19刘欢
文 / 刘欢
自2013年欧盟补充监测制度(Additional monitoring)实施以来,截至2017年9月底,欧洲药品管理局(EMA)更新并发布的补充监测目录中共纳入352种药品,其中包括154个含新活性物质药品和64个新生物制品(见表1)。研究欧盟补充监测制度对完善我国重点监测制度大有裨益。
补充监测基于安全的考虑
EMA发布补充监测目录是基于公众用药安全的考虑。所有药品获得许可的原则是该药品的疗效大于该药品所存在的潜在风险。要决定是否为一种药品授予上市许可,需要对该药品开发期间所做临床试验的数据进行评估。但是,对于一些极少出现或者在很长时间后才出现的不良反应(ADR),只有在药品被更广泛的人群使用和/或在长期使用之后,才可能会变得很明显。另外,药品风险获益评估的前提条件可能与日常医疗实践也有区别,例如临床试验可能会排除一些患有多种合并症或者合并用药的患者。因此,药品被放到市场之后,会在更大范围的人群中使用,这就需要上市许可持有人(MAH)和主管机构对药品进行持续监测,以发现新出现的信息,并评估信息是否对药品风险效益平衡产生影响。而对于一些特定药品,需要加强其上市后数据的收集,以确保及时发现并确定新的安全性风险,立即采取应对措施。为此,2010年,欧盟药物警戒法规中引入了补充监测的概念,2012年EMA又对药物警戒法规进行更新。为了新法规更加顺利的实施,EMA制定《药物警戒实践指南》(Guideline on good pharmacovigilance practices,GVP)作为欧盟药物警戒工作的新准则。2013年4月25日,GVP模块Ⅹ正式发布并实施,在该模块中提供了详细的补充监测具体操作指导。
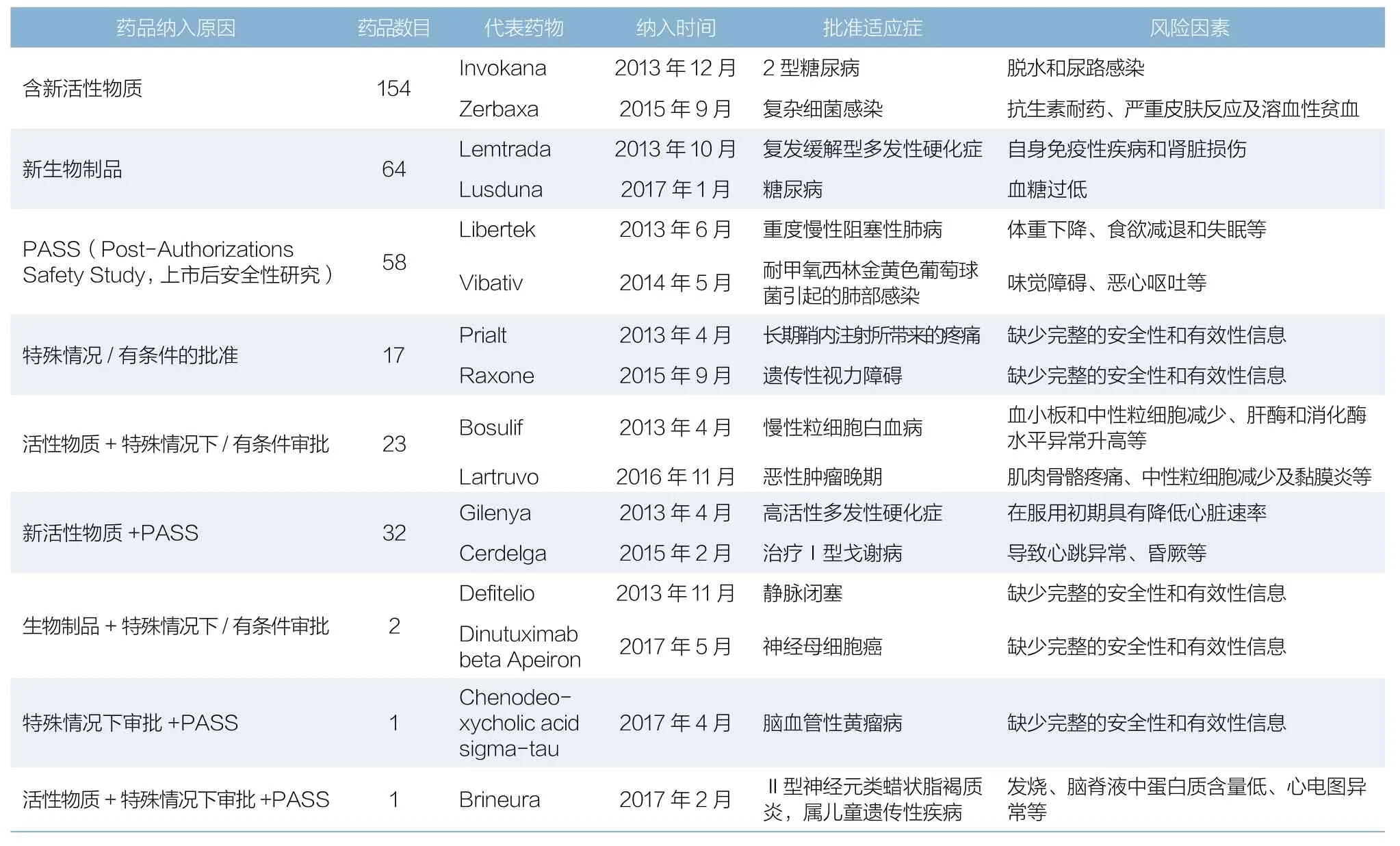
表1 EMA于2017年9月发布的补充监测药品目录
补充监测的理念主要源于提高以下两类产品不良反应报告比例的需要:一是新获得上市许可的药品,其安全性可能尚未得到完全确定;二是出现新安全性问题的药品,需要进一步确认其安全性。EMA及各成员国共同制定、维护和公布受补充监测的药品目录,及时公开目录中药品信息。对于目录中的药品,在其产品特征摘要中,加注黑色等边倒三角形,并同时在产品特征摘要和包装说明书中加注解释性说明,强化用药人群的识别等。鼓励医务人员和患者自发报告不良反应(ADR),以便顺利开展上市后不良反应的监测和评估,确保及时发现新的用药安全隐患并有效避免。
药品受补充监测不等于无效
已上市药品因为当前药品信息的不足,例如该药品刚获上市许可或缺少长期使用/更广泛人群使用信息,需要对其进行更加集中和频繁的监测,但并不意味着这类药品是无效的。
补充监测目录药品的纳入标准分为强制纳入药品及可选纳入药品两大类(见表2)。

表2 补充监测目录药品的纳入标准

欧盟补充监测制度对我国的启示
一般来说,对于2011年1月1日以后批准的含有新活性物质的药品及所有生物制品,初始监测周期为获上市许可之后的5年。对于其他纳进监测目录的药品,监测周期根据其数据收集和评价报告等工作完成情况而确定。
欧盟补充监测制度对我国的启示
2011年7月1日起我国施行的《药品不良反应报告和监测管理办法》(原卫生部令第81号)首次引入重点监测的概念——重点监测,即为进一步了解药品的临床使用和不良反应发生情况,研究不良反应的发生特征、严重程度、发生率等所开展的药品安全性监测活动。《办法》还限定重点监测药品范围,包括新药监测期内或首次进口五年内的药品、监管机构要求或生产企业主动开展重点监测的特定药品。2013年3月25日,原国家食品药品监督管理局发布《关于推动生产企业开展药品重点监测工作的通知(征求意见稿)》,并附《生产企业药品重点监测指南》,进一步推动了重点监测工作的发展。
借鉴欧盟药品补充监测制度,笔者建议继续完善我国重点监测制度。首先,要限定重点监测药品的具体范围。《药品不良反应报告和监测管理办法》中并没有限定特定药品的具体范围。其次,要建立并发布重点监测药品目录,并对目录中药品进行定期审查和更新(目前我国确实有部分地区发布了重点监控目录,但主要和药品招标采购有关,并不是基于药品潜在使用风险的考虑)。笔者认为,在将一种药品纳入重点监测目录时,应进行两方面的充分考量,一是是否能够提高人们安全有效地使用药品的意识,二是能否为该药品的安全性和有效性评估提供更多地信息。最后,在重点监测药品的说明书内添加特殊标志及解释性说明,要求使医务人员及患者容易辨识处于重点监测状态的药品,鼓励他们报告该类药品的不良反应。与此同时,要求医务人员与患者加强沟通,避免引起患者对使用该类药品的恐慌。
