团头鲂3个选育群体的选择压力分析
2017-12-18唐首杰王成辉张飞明张友良谢志强
唐首杰,毕 详,王成辉,张飞明,张友良,谢志强
( 1.上海海洋大学 农业部淡水水产种质资源重点实验室, 上海 201306;2.上海市松江区水产良种场,上海 201616 )
团头鲂3个选育群体的选择压力分析
唐首杰1,毕 详2,王成辉1,张飞明2,张友良2,谢志强2
( 1.上海海洋大学 农业部淡水水产种质资源重点实验室, 上海 201306;2.上海市松江区水产良种场,上海 201616 )
团头鲂选育群体基因组选择信号的检测有助于研究人工选育条件下基因组的进化机制,为团头鲂的进一步遗传改良提供依据。以团头鲂“浦江1号”选育奠基群体(F0)为对照组,以3个团头鲂选育群体为试验组,采用经典的Ewens-Watterson中性检验和基于3种算法(岛屿模型、分级岛屿模型、贝叶斯似然法)的FST-离群值点检验,在14个转录组微卫星位点上进行选择信号检测。结果显示,F0群体中所有位点均为中性位点,选育群体A在Mac927位点和Mac158位点受到了正向选择压力,选育群体B在Mac158位点受到了正向选择压力,选育群体C在Mac927位点和Mac158位点受到了正向选择压力。由此可见,自1985年起的连续世代人工选育已在团头鲂基因组中留下了可检测到的选择信号。3个选育群体均在Mac158位点检测到选择信号,表明3个选育群体受到的人工选择方向比较接近。
团头鲂;选育群体;选择压力
突变、遗传漂变、选择和基因流是自然生物进化的主要动力[1],对于大多数鱼类人工选育群体而言,在特定养殖环境下的人工选择是其进化的主要动力。鱼类的遗传改良通常会使群体中控制某些目标性状的等位基因频率不断提高[2],而这些等位基因频率提高的速度取决于人工选择的强度和精确度[3]。根据群体遗传学理论[4],受到选择作用的位点周围的中性位点会受遗传牵连效应的影响而出现基因频率的迅速提高[5],这是选择作用在基因组上留下的明显特征,通常被称为选择信号。已有的研究表明,长期的人工选育已在某些鱼类的基因组中留下了选择信号。Gutierrez等[6]利用单核苷酸多态性标记技术在大西洋鲑(Salmosalar)选育群体基因组中检测到44个与生长、驯化相关的选择信号。Xia等[7]通过基因组重测序技术在“吉富”品系尼罗罗非鱼(Oreochromisniloticus)基因组中检测到243个选择信号。
团头鲂(Megalobramaamblycephala)“浦江1号”是上海海洋大学经过六代高强度系统选育后获得的养殖性能优良的首例草食性鱼类选育良种[8]。本研究室在“浦江1号”良种(F10)的基础上,基于配套系育种理念[9-10],以不同选育目标分别建立了3个团头鲂选育群体,并对这3个选育群体进行了连续多代的选育纯化,为查明团头鲂选育群体中的选择信号,笔者进行了检测和分析,从而有助于了解人工选育条件下团头鲂基因组的进化机制。
近期,基于微卫星标记的FST-离群值点检测方法已被广泛应用于鱼类[如大西洋鲑[11]、鲱形白鲑(Coregonusclupeaformis)[12]、大西洋鳕(Gadusmorhua)[13]等]选择信号检测研究中。本研究以团头鲂“浦江1号”选育奠基群体为对照组,通过扫描14个高度多态的转录组微卫星位点来检测团头鲂3个选育群体基因组中受人工选择的印迹,旨在了解人工选育对团头鲂基因组产生的影响,为团头鲂选育群体进一步遗传改良提供依据,也为人工选育条件下团头鲂群体受到选择的遗传机制研究提供参考。
1 材料与方法
1.1 试验材料
对照组为团头鲂“浦江1号”选育奠基群体(以下简称F0),系1985年淤泥湖团头鲂原种在上海海洋大学鱼类种质研究试验站自繁的后代;试验组为3个团头鲂选育群体,其来源和性质如下。选育群体A(以下简称A):团头鲂“浦江1号”选育系F10;选育群体B(以下简称B):以人工诱导异源四倍体群体[团头鲂“浦江1号”♀×三角鲂(M.terminalis)♂][14]为奠基群体,经群体选育的第3代(F3);选育群体C(以下简称C):以团头鲂连续两代人工诱导减数分裂雌核发育群体[15]为奠基群体,经群体选育的第3代(F3)。对照组群体采自上海海洋大学鱼类种质研究试验站,试验组群体均采自上海市松江区水产良种场。每群体采样30尾,剪取每尾试验鱼的尾鳍,编号后于95%酒精中保存备用。
1.2 试验方法
1.2.1 基因组DNA提取
基因组DNA提取采用常规的“酚—氯仿”方法进行[16]。
1.2.2 微卫星引物的筛选和多态性引物的PCR扩增
41对微卫星引物:20对引物参照文献[17],15对引物参照文献[18],另有6对引物参照文献[19]。所有引物均由上海生工生物工程有限公司合成。
用41对引物进行PCR扩增,PCR扩增反应体积为10 μL,包括:正向引物0.5 μL(5 μmol/L),反向引物0.5 μL(5 μmol/L),2×PCR Reagent 5 μL,DNA模板1 μL,无菌水3 μL。PCR反应程序为:94 ℃预变性5 min,接下来进行35个循环,每个循环包括94 ℃变性30 s,退火45 s(各对引物退火温度见文献[17-19]),72 ℃延伸1 min;最后一次循环结束后在72 ℃延伸10 min。PCR产物经8.0%非变性聚丙烯酰胺凝胶电泳,银染法检测[16]。根据电泳结果中多态性的有无和扩增条带的清晰度来筛选引物,筛选其中多态性高且扩增清晰稳定的引物作为后续试验的引物。
经过筛选得到14对高度多态的微卫星引物(表1),以所有群体的基因组DNA为模板,用这14对微卫星引物进行PCR扩增,PCR扩增反应体系和程序同前。
1.2.3 毛细管电泳分析
采用QIAxcel全自动毛细管核酸分析系统(QIAGEN公司)对所有样本的PCR产物进行毛细管电泳分析,分析参数设置参照仪器使用说明书。利用两种DNA分子量标准,分别为pBR322DNA/MspⅠ和Alignment Marker(15 bp和500 bp),对PCR产物进行分子量估算。
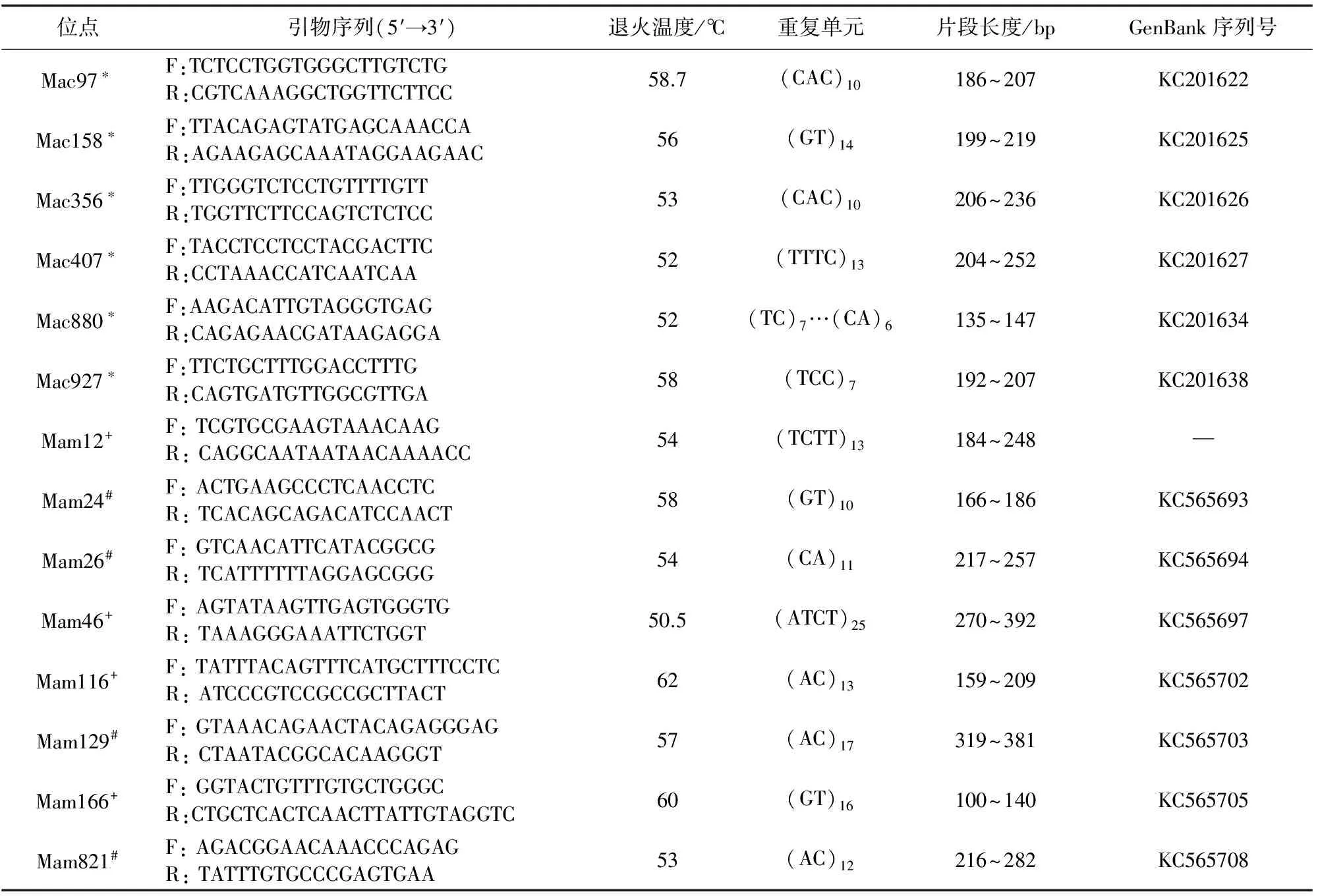
表1 14个团头鲂微卫星标记的特征
注:*, 表示该位点来源于文献[18]; #, 表示该位点来源于文献[18];+, 表示该位点来源于文献[19].
1.2.4 数据统计
首先利用 MICRO-CHECKER version 2.2.3软件[20]对得到的微卫星数据进行无效等位基因检测。随后将所有群体每个样本在14个微卫星位点上的基因型数据(等位基因片段)按CREATE软件[21]可识别的格式输入Microsoft Excel文件中,用CREATE软件将此数据格式文件分别转换为ARLEQUIN version 3.5.2.2软件[22]、FSTAT version 2.9.3软件[23]和GENEPOP version 4.0软件[24]可识别的格式文件。利用PGDSpider version 2.0.5.1软件[25]将ARLEQUIN version 3.5.2.2软件[22]可识别的格式文件转换成BayeScan version 2.1软件[26]可识别的格式文件。
采用FSTAT version 2.9.3软件[23]对所选用的14个微卫星位点进行连锁不平衡显著性检验,以确定位点间是否为独立遗传。
采用两种方法进行选择压力位点检测,一种方法基于Ewens-Watterson纯合度检验[27-28],通过对微卫星位点进行纯合度(F)过度或缺失检验,来判断该位点是否偏离中性模型。采用Manly[29]的算法对每个位点进行Ewens-Watterson纯合度检验,若某位点观测纯合度位于期望纯合度的95%置信区间内,则该位点符合中性模型;若某位点观测纯合度超出了期望纯合度的95%置信区间,则该位点偏离中性模型,即受到了选择作用。以上运算在POPGENE软件[30]中完成,参数设置为:模拟1000次,显著性水平定在α=0.05。
另一种方法基于FST-离群值检验[31],采用3种算法来进行FST-离群值检验,以检测受到正向选择的位点。第一种算法基于Beaumont等[32]提出的岛屿模型,使用LOSITAN version 1.44软件[33]构建遗传分化指数(FST)与杂合度的条件联合分布曲线图,超出99%置信区间上限的位点为受到强烈正向选择的位点。LOSITAN软件的参数设置为:选择逐步突变模型,模拟50000次,置信区间为99%。
第二种算法基于Excoffier等[34]提出的分级岛屿模型,在ARLEQUIN version 3.5.2.2软件[22]上进行运算,由R函数(https://www.R-project.org/)构建遗传分化指数(FST)与杂合度的条件联合分布曲线图,超出99%置信区间上限的位点为受到强烈正向选择的位点。参数设置为:选择逐步突变模型(SMM),模拟20000次,置信区间为90%,95%和99%。
第三种算法为贝叶斯似然法,通过可逆跳跃马尔科夫链蒙特卡罗算法获得后验分布[35],参数设置为burn-in= 50 000次;chain length =500 000次。在BayeScan version 2.1软件[26]上完成运算。根据Jeffrey[36]的方法,贝叶斯因子(BF)>10[或Log10(BF)>1]时,表明某位点受到了强烈的正向选择。
以上3种算法所检测到的所有受正向选择的位点中,至少被2种算法同时检测到的位点可判定为受到强烈正向选择的位点。
2 结 果
2.1 毛细管电泳及基因分型结果
毛细管电泳能直接从峰形图上判读出纯合子(图1)和杂合子(图2),并通过与DNA标准分子量进行比对后得出每个峰对应的片段长度。引物Mac407在A群体中的第1号样本的电泳结果见图1,引物Mac407在A群体中的第2号样本的电泳结果见图2。引物Mac407在A群体第1~11号样本的电泳结果见图3。
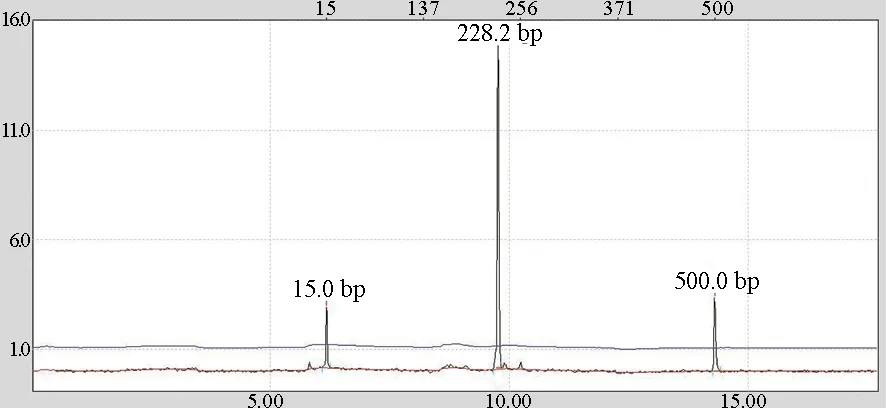
图1 引物Mac407在A群体中的第1号样本的电泳结果
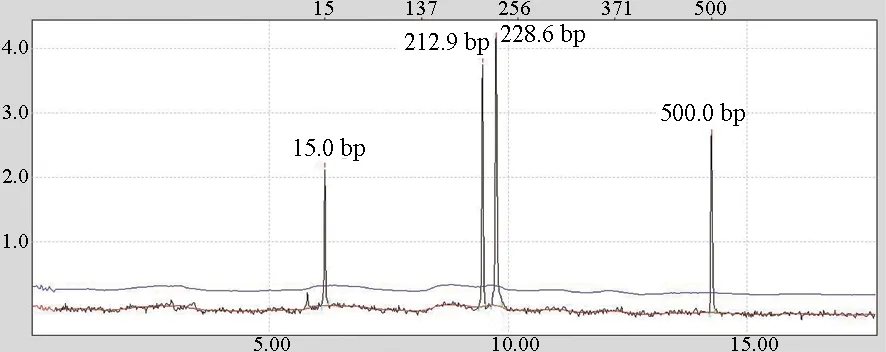
图2 引物Mac407在A群体中的第2号样本的电泳结果
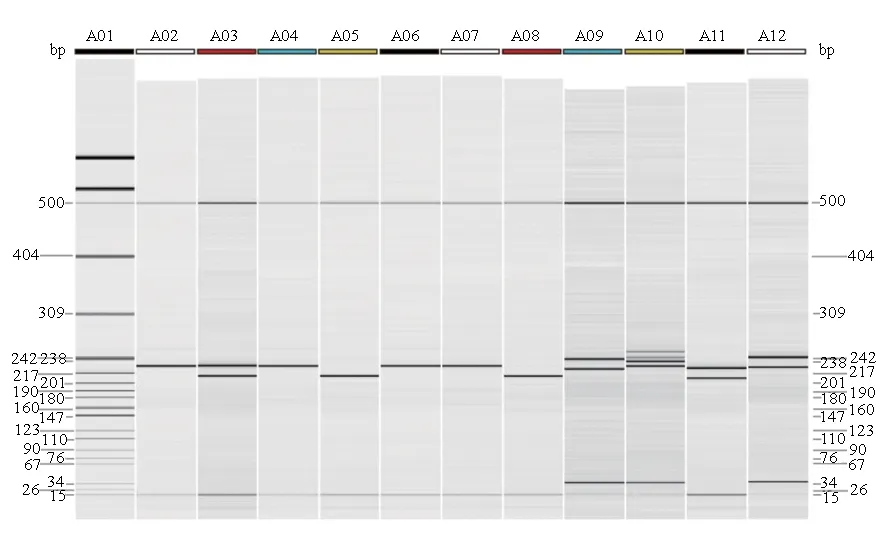
图3 引物Mac407在A群体第1-11号样本的电泳结果A01: pBR322DNA/MspⅠ, A02-A12: 第1~11号样本.
采用 MICRO-CHECKER version 2.2.3软件对4个群体的每个微卫星位点进行无效等位基因检测,发现所有位点均不存在无效等位基因。经成对位点间的连锁不平衡检验,发现各位点间连锁不平衡检验不显著(P>0.05)。说明本研究使用的14个微卫星位点均是独立遗传的,因此保留所有位点进行后续分析。
2.2 选择压力分析结果
2.2.1 Ewens-Watterson中性检验结果
对14个微卫星位点进行Ewens-Watterson中性测试,在1000次重复模拟条件下,在95%置信区间内检测到的选择压力位点见表2。其中,F0群体和B群体中所有位点的观测纯合度均处于期望纯合度的95%置信区间内,即所有位点均为中性位点。A群体和C群体在Mac927位点的观测纯合度低于期望纯合度的95%置信区间下限,即Mac927位点偏离了中性模型,可能为受到选择压力的位点。
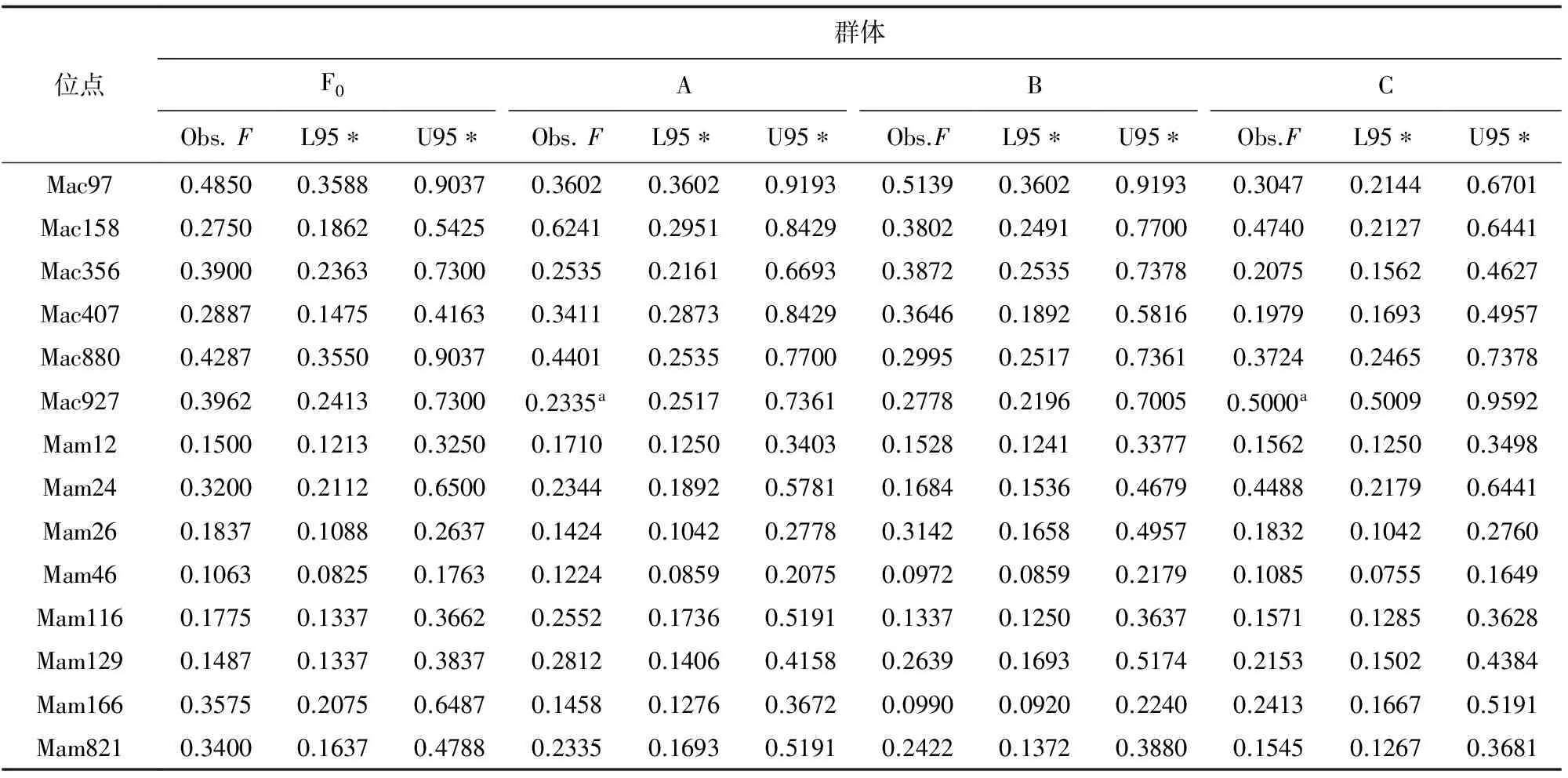
表2 4个团头鲂群体在14个微卫星位点的Ewens-Watterson中性检验结果
注:Obs.F,观测纯合度;L95,95%置信区间下限;U95,95%置信区间上限;a,偏离中性模型. *,此统计是通过1000次模拟计算而得到的结果.
2.2.2FST-离群值点检测结果
在对14个微卫星位点进行选择压力分析前,先将受试群体进行了分组,将所有群体按其性质分为以下两种分组模式:第一种模式,将所有群体归为1组,对所有群体进行分析;第二种模式,将3个选育群体归为1组,只对3个选育群体进行分析。分别应用3种软件(LOSITAN、ARLEQUIN、BAYESCAN)对14个微卫星位点进行选择压力分析的结果显示,在第一种模式下,LOSITAN软件在Mac158和Mac880位点检测到显著的正向选择压力(图4a),ARLEQUIN软件在Mac158位点检测到显著的正向选择压力(图5a),BAYESCAN软件在Mam26和Mam46位点检测到显著的正向选择压力(图6a)。在第二种模式下,LOSITAN软件在Mac158、Mac880、Mac927和Mam821位点检测到显著的正向选择压力(图4b),ARLEQUIN软件在Mac158位点检测到显著的正向选择压力(图5b),BAYESCAN软件在Mam26位点检测到显著的正向选择压力(图6b)。综上所述,因3种软件检测到的选择压力位点不一致,为增加检测结果的可靠性,在本研究中,被两种软件同时检测到的位点可判定为受到正向选择的位点(表3),因此,在第一种模式下,所有群体在Mac158位点受到了显著的正向选择压力;在第二种模式下,3个选育群体在Mac158位点受到了显著的正向选择压力。
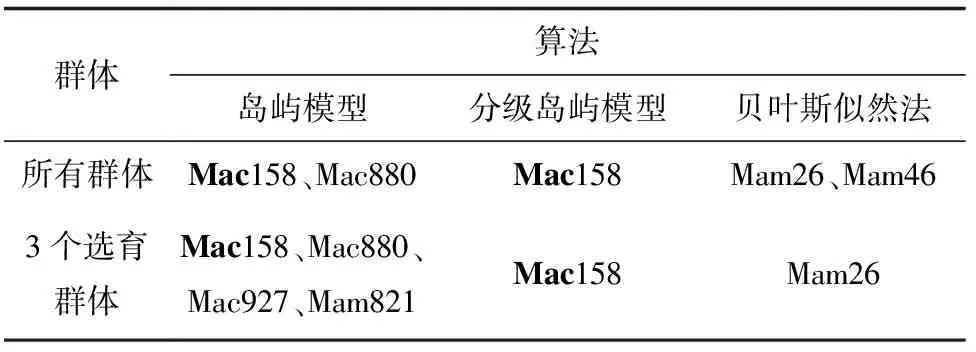
表3 3种FST-离群值算法检测到的受正向选择位点统计表
注:加粗的位点为至少被两种算法同时检测到受正向选择的位点.
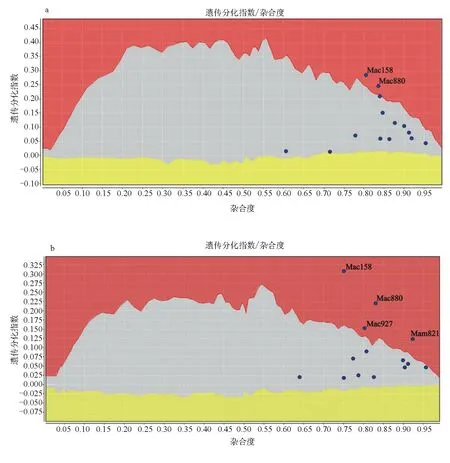
图4 LOSITAN软件在2种模式下的选择压力位点分析结果a图表示对所有群体的检测结果;b图表示对3个团头鲂选育群体的检测结果.红色区域为超出99%置信区间上限的区域,灰色区域为处于99%置信区间以内的区域,黄色区域为超出99%置信区间下限的区域,红色区域内的点为受正向选择的位点,灰色区域内的点为中性进化位点.
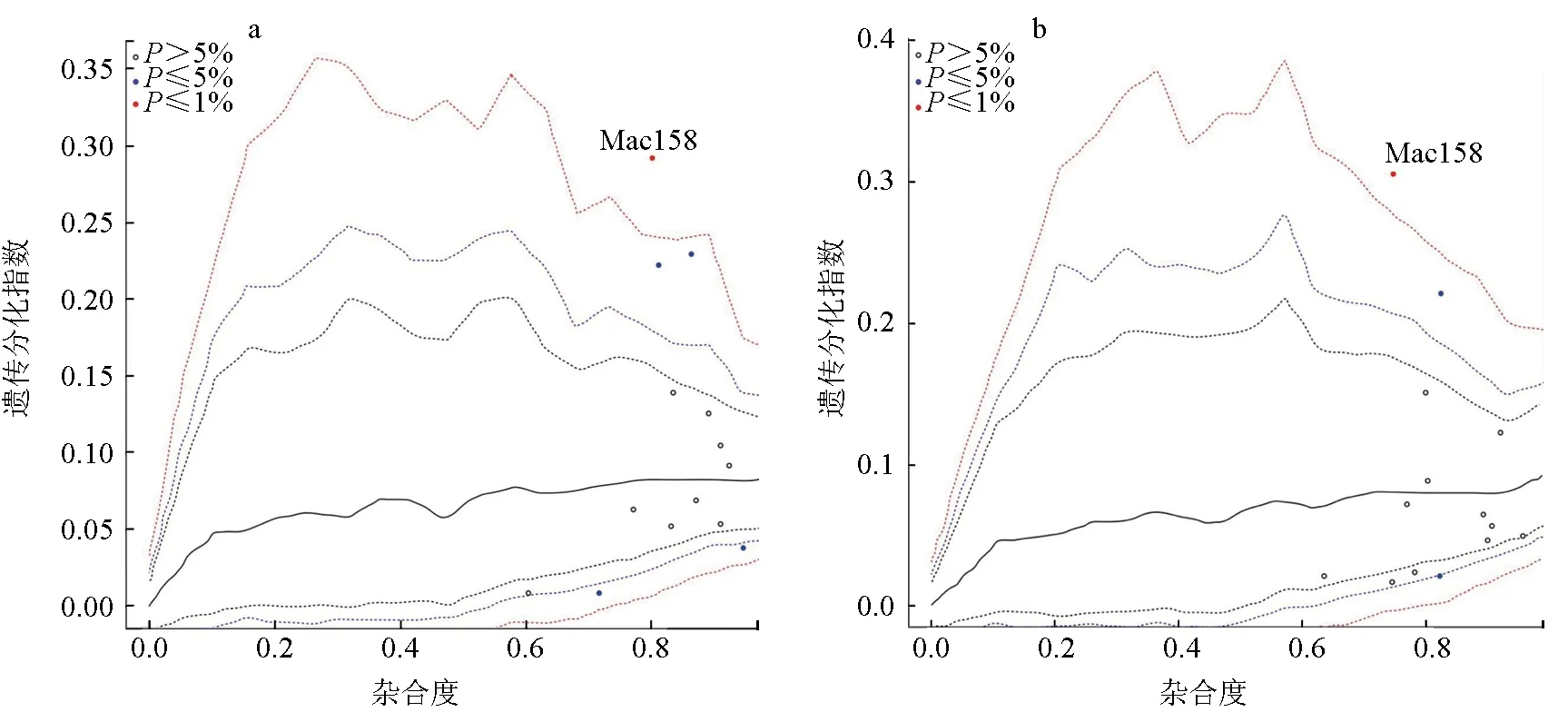
图5 ARLEQUIN软件在两种模式下的选择压力位点分析结果a图表示对所有群体的检测结果;b图表示对3个团头鲂选育群体的检测结果.红色虚线表示99%置信度水平,蓝色虚线表示95%置信度水平,黑色虚线表示90%置信度水平,黑色实线表示50%置信度水平,红色实心点表示超出99%置信度水平的位点,蓝色实心点表示处在95%~99%置信度内的位点,空心点表示处于95%置信度以内的位点.
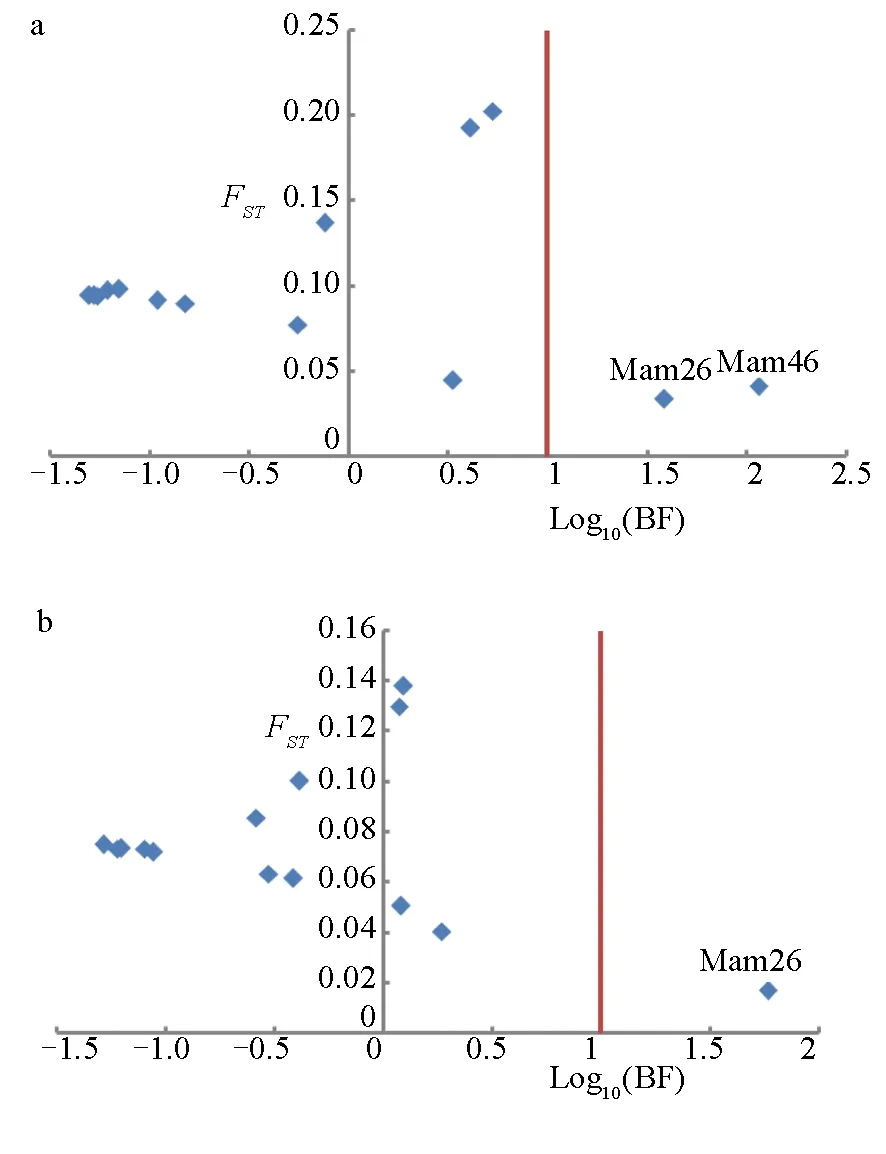
图6 BayeScan软件在两种模式下的选择压力位点分析结果a图表示对所有群体的检测结果;b图表示对3个团头鲂选育群体的检测结果. 红色实线右侧的正方形图标为受到正向选择的位点.
3 讨 论
人工选择在动植物品种培育和经济性状遗传改良上发挥着举足轻重的作用。鱼类的人工驯养历史非常短暂[37]。如大西洋鲑的驯养和人工选育始于20世纪70年代[38]。尽管如此,短暂而高强度的人工选择已在大西洋鲑[39]、大菱鲆(Scophthalmusmaximus)[40]等的基因组中留下了选择烙印。团头鲂的驯化和人工养殖始于20世纪60年代[41],其人工选育始于1985年,历经16年(六代)高强度系统选育后,选育系第六代(F6)的生长速度已比原种提高30%,生长优势明显,遗传性状稳定[8]。3个选育群体(A、B、C)是在选育系F6基础上建立的3个新品系,并持续选育至今,因此,这3个选育群体均经历了长达30年的多代人工选育。选择压力位点检测结果表明,人工选育已在团头鲂3个选育群体基因组中留下了选择信号,其中,A群体和C群体同时在2个位点(Mac927和Mac158)检测到选择信号,B群体只在1个位点(Mac158)检测到选择信号。值得注意的是,3个选育群体均在同一个位点(Mac158)检测到了选择信号,表明它们受到的人工选择方向比较接近。这可能是因为这3个选育群体建立的时间较晚,且各自经历的选育世代数较少(3~4代)而造成的。此外,位点Mac927和Mac158均来自团头鲂转录组数据库[19],表明它们可能是功能基因的一部分,也可能与生长等表型性状相关。而要了解这些位点在团头鲂基因组中的具体位置和功能,仍需开展进一步的深入研究。
4 结 论
本研究中使用的两种选择压力检测方法分别基于不同的群体遗传学假设,很难说哪一种方法更能反映微卫星位点受到选择的真实情况,笔者认为应综合考虑两种方法的检测结果,即F0群体中所有位点均为中性位点,A群体在Mac927位点和Mac158位点受到了正向选择压力,B群体在Mac158位点受到了正向选择压力,C群体在Mac927位点和Mac158位点受到了正向选择压力。
[1] 沈银柱, 黄占景. 进化生物学[M]. 北京:高等教育出版社, 2012:139-147.
[2] 楼允东. 鱼类育种学[M]. 北京:中国农业出版社, 2001:10-14.
[3] Falconer D S. Introduction to Quantitative Genetics [M]. 3rd Ed. England: Longman Scientific & Technical, 1989:201-206.
[4] Nielsen R. Molecular signatures of natural selection [J]. Annual Review of Genetics, 2005(39):197-218.
[5] Smith J M, Haigh J. The hitch-hiking effect of a favourable gene[J]. Genetics Research, 1974, 23(1):23-35.
[6] Gutierrez A P, Yáez J M, Davidson W S. Evidence of recent signatures of selection during domestication in an Atlantic salmon population[J]. Marine Genomics, 2015(26):41-50.
[7] Xia J H, Bai Z Y, Meng Z N, et al. Signatures of selection in tilapia revealed by whole genomere sequencing[J]. Scientific Reports, 2015, 5(5):1-10.
[8] Li S F, Cai W Q. Genetic improvement of the herbivorous blunt snout bream (Megalobramaamblycephala)[J]. NAGA, 2003, 26(1):20-23.
[9] 张沅. 家畜育种学[M].北京:中国农业出版社, 2007:230-254.
[10] Wang C H, Wang J, Xiang S P, et al. Parental genetic effects evaluation of growth-related traits of red common carp in China[J]. Fisheries Science, 2009, 75(5):1301-1305.
[11] Vasemägi A, Nilsson J, Primmer C R. Express sequence Tag-linked microsatellite as a source for gene-associated polymorphisms for detecting signatures of selection in Atlantic salmon (SalmosalarL.)[J]. Molecular Biology and Evolution, 2005, 22(4):1067-1076.
[12] Campbell D, Bernatchez L. Genetic scan using AFLP markers as a means to assess the role of directional selection in the divergence of sympatric whitefish ecotypes[J]. Molecular Biology and Evolution, 2004, 21(5):945-956.
[13] Nielsen E E, Hemmer-Hansen J, Poulsen N A, et al. Genomic signatures of local directional selection in a high gene flow marine organism:the Atlantic cod (Gadusmorhua)[J].BMC Evolutionary Biology, 2009(9):276.
[14] 邹曙明.团头鲂人工同源和异源四倍体的繁育群体建立及其不同倍性后代的生物学特征分析[D].上海:上海水产大学,2004.
[15] 李思发, 杨怀宇, 邹曙明.快速近交对团头鲂遗传结构的影响和近交效应的估算[J].水产学报, 2005,29(2):161-165.
[16] Sambrook J, Russell D W. 黄培堂, 王嘉玺, 朱厚础,等译.分子克隆实验指南[M].3版. 北京:科学出版社, 2002:463-470,1730-1732.
[17] Luo W, Deng W, Yi S K, et al. Characterization of 20 polymorphic microsatellites for blunt snout bream (Megalobramaamblycephala) from EST sequences[J]. Conservation Genetics Resources, 2013, 5(2): 499-501.
[18] 罗伟. 团头鲂EST-SSR的开发及在育种中的应用[D]. 武汉:华中农业大学, 2014.
[19] Gao Z, Luo W, Liu H, et al. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobramaamblycephala)[J]. PLoS One, 2012, 7(8):42637.
[20] Vanoosterhout C, Hutchinson W F, Wills D P M, et al. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data[J]. Molecular Ecology Notes, 2004, 4(3):535-538.
[21] Coombs J A, Letcher B H, Nislow K H. CREATE: a software to create input files from diploid genotypic data for 52 genetic software programs[J]. Molecular Ecology Resources, 2008, 8(3):578-580.
[22] Excoffier L, Lischer H E L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows[J]. Molecular Ecology Resources, 2010, 10(3):564-567.
[23] Goudet J. FSTAT: a Program to Estimate and Test Gene Diversities and Fixation Indices (version 2.9.3)[EB/OL]. 2012, [2016-04-21] http://www.unil.ch/dee/home/menuinst/softwares--dataset/softwares/fstat.html.
[24] Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism[J]. Journal of Heredity, 1995, 86(3):248-249.
[25] Lischer H E L, Excoffier L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs[J]. Bioinformatics, 2012, 28(2):298-299.
[26] Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective[J]. Genetics, 2008, 180(2):977-993.
[27] Ewens W J. The sampling theory of selectively neutral alleles[J]. Theoretical Population Biology, 1972, 3(1):87-112.
[28] Watterson G. The homozygosity test of neutrality[J]. Genetics, 1978, 88(2):405-417.
[29] Manly B F J. The statistics of natural selection on animal populations[M]. London:Chapman and Hall, 1985:272-285.
[30] Yeh F C, Boyle T J B. Population genetic analysis of co-dominant and dominant markers and quantitative traits[J]. Belgian Journal of Botany,1997, 129(2):157.
[31] Lewontin R, Krakauer J. Distribution of gene frequency as a test of theory of selective neutrality of polymorphisms[J].Genetics, 1973, 74(1):175-195.
[32] Beaumont M A, Nichols R A. Evaluating loci for use in the genetic analysis of population structure[J]. Proceedings of the Royal Society of London B—Biological Sciences, 1996, 263(1377):1619-1626.
[33] Antao T, Lopes A, Lopes R J, et al. LOSITAN: workbench to detect molecular adaptation based on a Fst-outlier method[J]. BMC Bioinformatics, 2008(9):323.
[34] Excoffier L, Hofer T, Foll M. Detecting loci under selection in a hierarchically structured population[J]. Heredity, 2009, 103(4):285-298.
[35] Green P J. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination[J]. Biometrika, 1995, 82(4):711-732.
[36] Jeffrey H. Some tests of significance, treated by the theory of probability[J]. Proceedings of the Cambridge Philosophy Society, 1935, 31(2):203-222.
[37] Mignon-Grasteau S, Boissy A, Bouix J, et al. Genetics of adaptation and domestication in livestock[J]. Livestock Production Science, 2005, 93(1):3-14.
[38] Gjedrem T. Genetic improvement for the development of efficient global aquaculture: a personal opinion review[J]. Aquaculture, 2012(344/349):12-22.
[39] Martinez V, Dettleff P, Lopez P, et al. Assessing footprints of selection in commercial Atlantic salmon populations using microsatellite data[J]. Animal Genetics, 2012, 44(2):223-226.
[40] Vilas R, Vandamme S G, Vera M, et al. A genome scan for candidate genes involved in the adaptation of turbot (Scophthalmusmaximus)[J]. Marine Genomics, 2015(23):77-86.
[41] 柯鸿文. 一种优良淡水鱼─团头鲂(Megalobramaamblycephala)的繁殖和饲养[J]. 水生生物学集刊, 1975,5(3):293-314.
SelectivePressureonThreeSelectiveBreedingPopulationsofBluntSnoutBreamMegalobramaamblycephala
TANG Shoujie1, BI Xiang2, WANG Chenghui1, ZHANG Feiming2, ZHANG Youliang2, XIE Zhiqiang2
( 1.Key Laboratory of Freshwater Fishery Germplasm Resources, Ministry of Agriculture, Shanghai Ocean University, Shanghai 201306, China;2. Songjiang Aquatic Seed Breeding Farm, Shanghai 201616, China )
Identification of selection signatures in genomic regions of selective breeding populations of blunt snout bream (Megalobramaamblycephala) leads to contribute not only to the study of evolution mechanisms of genome under artificial selection, but also to provide the basis for further genetic improvement. A population genetic approach, using a classical Ewens-Watterson neutrality test and three algorithms (Island model, Hierarchical island model, and Bayesian likelihood method) ofFST-outlier detection methods, was employed to evaluate polymorphism in fourteen transcriptomic microsatellite loci in order to identify loci that may be candidates for selection amongst four populations (three selective breeding populations and one foundation population of “Pujiang 1” selected strain) of blunt snout bream.The results showed that no selection signature was detected in foundation population (F0).Two loci (Mac927 and Mac158) showed significant evidence of positive artificial selection in both selective breeding population A and population C. Only one loci (Mac158) experienced positive artificial selection in selective breeding population B. The findings indicated that detectable selection signatures were found in the genome of blunt snout bream by successive generations of artificial selection since 1985. The selection signature of Mac158 was detected in each selective breeding population, indicating that there was similar direction of artificial selection in the three selective breeding populations.
Megalobramaamblycephala; selective breeding population; selective pressure
10.16378/j.cnki.1003-1111.2017.03.005
S965.119
A
1003-1111(2017)03-0282-08
2016-04-21;
2016-07-07.
上海市科技兴农重点攻关项目[沪农科攻字(2013)第2-3号].
唐首杰(1981-),男,讲师,博士;研究方向:水产动物种质资源与种苗工程. E-mail:sjtang@shou.edu.cn.
