利用电穿孔法对狂犬病病毒SRV9 Ψ区缺失株病毒的拯救及鉴定
2017-12-15毛丽萍魏玉圆翟少华文兆海简子健
毛丽萍,魏玉圆,王 伟,翟少华,程 瑶,文兆海,简子健
(新疆农业大学动物医学学院,新疆乌鲁木齐 830052)
利用电穿孔法对狂犬病病毒SRV9 Ψ区缺失株病毒的拯救及鉴定
毛丽萍△,魏玉圆△,王 伟,翟少华,程 瑶,文兆海,简子健*
(新疆农业大学动物医学学院,新疆乌鲁木齐 830052)
探讨电穿孔转染法对SRV9 Ψ区缺失株重组病毒的拯救及鉴定。利用电穿孔细胞转染法,将纯化的Ψ区缺失株pcDNA-NPMG-L全长质粒和pcDNA-N、pcDNA-P、pcDNA-M、pcDNA-G和pcDNA-L辅助表达质粒共转染至BHK-21细胞中,并进行盲传,借助RT-PCR、间接免疫荧光试验、Western-blot及透射电子显微镜对重组病毒进行鉴定。结果显示,重组病毒能够产生绿色荧光,透射电子显微镜观察可见典型的弹状病毒,对重组毒株与亲本毒株G蛋白进行Western-blot验证,可获得相应目的条带。结果表明,通过电穿孔细胞转染法,能成功拯救出重组病毒SRV9 Ψ区缺失株,为病毒的拯救提供新的转染方法,并能有效提高转染效率,为研发新型狂犬病疫苗提供参考。
狂犬病病毒;电穿孔转染法; SRV9 Ψ区缺失株;拯救
狂犬病(Rabies)是由狂犬病病毒引起的一种人畜共患的中枢神经系统传染病,病死率高达100%[1]。该病毒为弹状病毒科狂犬病毒属的单股负链RNA病毒。基因组全长约12kb,由5个结构基因构成,从3′到5′端依次为N、P、M、G、L基因,各个基因间含有非编码的间隔序列。在G-L基因之间存在423个核苷酸构成的间隔序列为一段高度保守的区域,被称伪基因(简称Ψ区)。研究表明,缺失Ψ区域不会影响病毒在宿主细胞内的扩增,并证实Ψ区域具有表达外源基因的潜在能力[2]。
狂犬病病毒的拯救过程,需将重组全长cDNA和辅助质粒共同转染至细胞内,获得具有感染性并能够稳定遗传的RNA病毒。电穿孔细胞转染是利用瞬间产生高强度电场产生的电脉冲改变细胞膜的通透性,将外源基因导入细胞内的方法[3]。该方法简单、易行[4],且安全性好、转化效率高,但对细胞具有一定的损伤性[5-6]。
本研究利用已构建的狂犬病SRV9 Ψ区缺失株感染性cDNA,采用电转染法将重组质粒与辅助质粒共转染BHK-21细胞,拯救获得缺失Ψ区的狂犬病病毒SRV9株病毒,利用间接免疫荧光、RT-PCR、缺失株病毒液的透射电子显微镜观察等进行鉴定,为深入研究新型狂犬病疫苗提供新的思路与试验依据。
1 材料与方法
1.1 材料
1.1.1 细胞和质粒 叙利亚幼仓鼠肾细胞(BHK-21);表达绿色荧光蛋白质粒pMAX(德国,Lonza公司);狂犬病病毒SRV9 Ψ区缺失株感染性cDNA(pCDNA-NPMG-L )全长质粒,pcDNA-N、pcDNA-P、pcDNA-M、pcDNA-G和pcDNA-L辅助表达质粒均由新疆农业大学动物医学学院病理实验室构建并保存。
1.1.2 主要试剂 DNA标准DL 2 000、Prime STAR®Max DNA Polymerase购自TaKaRa公司;InvitrogenTMM-MLV Reverse Transcriptase购自Thermo Fisher Scientific公司;美国Hyclone胎牛(FBS)、MEM高糖培养基购自宝生物工程(大连)有限公司;狂犬病的鼠源性阳性血清购于长春军事兽医研究所;鼠抗RV GP高免血清、FITC标记山羊抗小鼠的荧光抗体和辣根过氧化物标记山羊抗小鼠的抗体均购北京博奥森公司。
1.2 方法
1.2.1 引物设计与合成 根据GenBank中已发布的SRV9株基因组序列(登录号:AF499686.2),利用Oligo 6.0软件设计检测SRV9 Ψ区缺失株感染性cDNA的鉴定引物,GLF:5′-GCAGACCCGTCTACCG TTTT-3′;GLR:5′-GACAATGGGGGTTCCTCTGG-3′,由上海生工生物工程股份有限公司合成。
1.2.2 SRV9 Ψ区缺失株电转染及扩增 待BHK-21细胞生长密度约为80% 时,消化收集并将细胞调至1.8×106个细胞,100 μL电转液悬浮(S1∶S2=1∶50),转入pMAX质粒(阳性对照)、重组缺失株全长质粒pcDNA-NPMG-L和辅助质粒pcDNA-N、pcDNA-P、pcDNA-M、pcDNA-G、pcDNA-L用量分别为1.0、2.5、1.5、0.8、0.5、0.6、0.5 μg。将电击杯放置于电转槽中,运行EN-138电转程序,室温静置10 min,置于37℃、体积分数为5% CO2培养。培养6 h后换液,37℃、体积分数为5% CO2培养,48 h后将培养皿置于荧光倒置显微镜下进行观测。待细胞单层铺满培养皿,盲传并反复冻融,离心收集上清,收获缺失株病毒液。
1.2.3 SRV9 Ψ区缺失株病毒的RT-PCR检测 取缺失毒株和标准毒株的病毒液提取总RNA,参照InvitrogenTMM-MLV Reverse Transcriptase试剂操作指南,进行cDNA第一链的合成。根据狂犬病病毒SRV9 Ψ区缺失株基因特点,设计用于对所拯救的狂犬病病毒SRV9缺失ψ区株病毒进行鉴定的引物GLF、GLR,以cDNA为模板,进行PCR扩增,10 g/L琼脂糖凝胶电泳进行验证。
1.2.4 SRV9 Ψ区缺失株病毒的间接免疫荧光检测 按1.8×106个/mL细胞密度传至6孔板。同步接种缺失株病毒液(70 μL/孔)及亲本毒株(阳性对照)与正常细胞(阴性对照),并加入维持液。37℃、体积分数为5% CO2培养箱培养72 h;弃去上清,预冷的800 mL/L丙酮,4℃固定12 h;1∶2 000稀释的狂犬病阳性血清为一抗,37℃感作2 h,在暗室中加入1∶200稀释的FITC标记山羊抗鼠为二抗,37℃感作30 min。在共聚焦显微下观察,对缺失株病毒进行检测。
1.2.5 SRV9 Ψ区缺失株病毒G蛋白的Western-blot检测 在缺失株的病毒液、原毒株病毒液及空白细胞体中加入150 μL蛋白抽提试剂(RIPA:蛋白抑制剂=100∶1),冰上孵育30 min,4℃、12 000 r/min离心10 min,滴加裂解液100 μL至相应的管中,分别加入等体积的2×蛋白上样缓冲液。PCR仪控温99℃ 8 min。12%SDS-PAGE电泳后转印至PVDF膜,膜封闭液4 ℃封闭过夜;1∶5 000稀释的鼠抗RV GP高免血清4 ℃过夜;1∶7 000 稀释的辣根过氧化物标记的山羊抗鼠二抗4 ℃过夜;洗膜加入ECL显色剂,在蛋白印迹扫描成像系统下记录试验结果。
1.2.6 SRV9 Ψ区缺失株病毒液的透射电子显微镜检测 将缺失株原毒株病毒液分别与狂犬病抗体反应后,吸取少量病毒液滴于附有Formvar膜的铜网上,静置2 min,用滤纸吸去多余液体,悬滴2%磷钨酸,静置2 min,用滤纸吸去染液,自然干燥后进行透射电子显微镜观察。
1.2.7 狂犬病病毒SRV9 Ψ区缺失株病毒感染细胞的透射级电子显微镜检测 分别取过滤缺失株病毒液、原SRV9株的病毒液,采用同步接毒法,将两种病毒分别感染BHK-21细胞。待细胞长满,消化收集置于1.5 mL离心管中,加1 mL的2.5%戊二醛,4℃固定24 h;PBS洗涤3次,30 min/次;将细胞样品包埋于40 g/L琼脂中,加入10 mL/L锇酸,4℃固定2 h;PBS洗涤3次,30 min/次;在30%、50%、70%、80%、90%、100%逐级脱水,每个浓度15 min;用无水丙酮对样品内的脱水剂进行置换;用包埋剂进行渗透,将包埋样品放入聚合器中进行聚合,温度和时间按照37℃ 12 h,45℃ 24 h,60℃ 48 h依次进行;聚合后,对制备包埋块进行修块制备超薄切片。切片用醋酸铀染色,自然干燥后在透射电子显微镜下观察拍照。
2 结果
2.1 转染效率
电转染48 h后,通过荧光倒置显微镜下对转染的pMAX质粒的BHK-21细胞进行观察,产生大量绿色荧光(图1 B),表明阳性pMAX质粒电转染效率较高;而未经转染pMAX质粒的对照组中BHK-21细胞未产生绿色荧光(图1 C)。
2.2 RT-PCR 检测
采用引物GLF、GLR,以重组全长cDNA质粒做阳性对照,取亲本SRV9病毒作平行对照,通过电转染拯救的狂犬病病毒SRV9 Ψ区缺失株进行RT-PCR检测。标准毒株片段为845 bp,缺失株目的片段为422 bp,大小趋于一致(图2)。
2.3 间接免疫荧光检测
将重组病毒进行间接免疫荧光检测。设置亲本SRV9毒株做阳性对照、未感染病毒的正常细胞做阴性对照、转染所有辅助质粒组的细胞及仅转染全长质粒组做平行对照。通过共聚焦显微镜观察发现,正常BHK-21细胞、仅转染辅助质粒组和转染全长基因表达质粒组均未见任何荧光(图3B-D)。转染的重组全长质粒和辅助质粒组与阳性对照的亲本毒株组,均产生大量绿色荧光(图3E-F)。由此说明已成功拯救出狂犬病病毒SRV9 Ψ区缺失株。
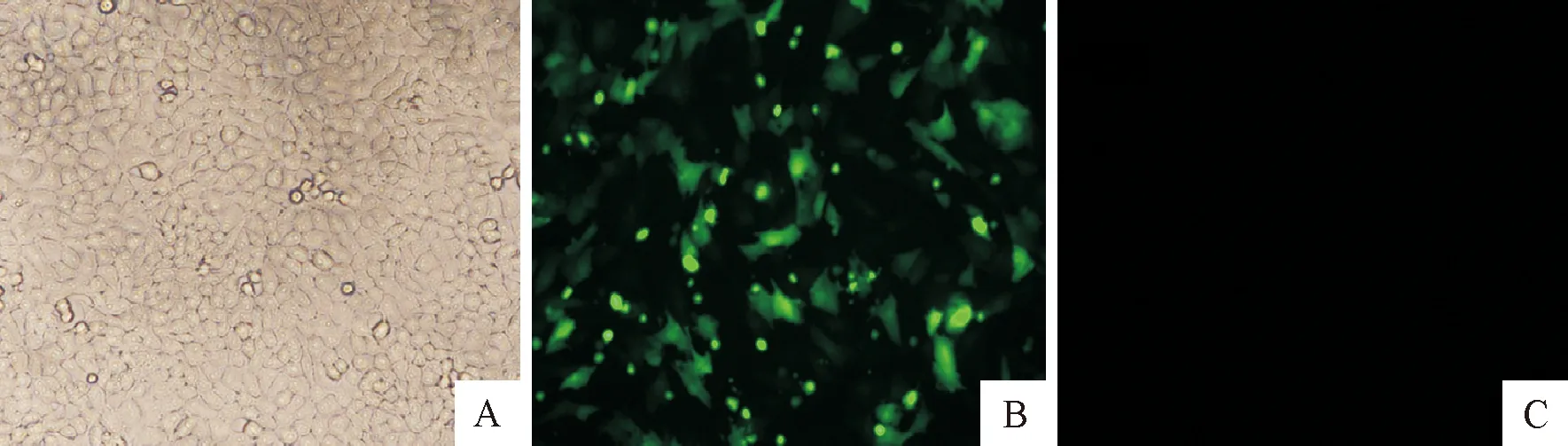
A.正常BHK-21细胞 B.转染pMAX荧光图 C.未转染pMAX荧光图A.Normal BHK-21 cells; B.Images of fluorescence with transfection pMAX; C.Images of fluorescence without transfection pMAX
M.DNA 标准 DL 2 000;1.阴性对照;2.全长质粒阳性对照(422 bp);3.重组病毒RT-PCR检测(422 bp);4.亲本毒株RT-PCR 检测(845 bp);5.转染辅助质粒;6.转染全长质粒;7.转染空白细胞
M.DNA Marker DL 2 000;1.Negative control;2.Positive control for plasmids of entire length(422 bp); 3.Detection of recombinant virus by RT-PCR(422 bp); 4.Detection of parent strain by RT-PCR; 5.Auxiliary plasmid transfection; 6.Transfection of entire length plasmid; 7.Transfection of blank cells
图2重组病毒的RT-PCR检测
Fig.2 Deteciont of recombinant virus by RT-PCR
2.4 Western blot检测
取狂犬病病毒SRV9 Ψ区缺失株病毒液、亲本SRV9株的病毒液与空白细胞体,进行G蛋白的Western blot检测。结果显示,拯救毒株与亲本毒株能产生大小相近的G蛋白条带,而空白对照均无任何条带(图4)。
2.5 病毒液的电镜检测
将狂犬病病毒SRV9 Ψ区缺失株病毒液和亲本SRV9株的病毒液分别与狂犬病阳性血清反应形成免疫复合物。负染制样后透射显微镜下观察,在8万倍下可观察到子弹状的典型狂犬病病毒粒子(图5)。
2.6 病毒颗粒电镜检测
将狂犬病病毒SRV9 Ψ区缺失株病毒液和亲本SRV9株的病毒液同步接毒去感染细胞,收集细胞制样后,在3万倍和5万倍透射电子显微镜下可见大量聚集弹状的狂犬病病毒形态的颗粒(图6)。
3 讨论
狂犬病病毒SRV9株是SAD株在BHK-21细胞系上通过空斑和动物脑内接种得到的弱毒疫苗株,具有较好免疫原性,并且对多种动物无致病性,对机体的保护率可达100%,优于SAD株[7-9]。狂犬病病毒基因组中,位于G-L基因之间的间隔区是一个高度保守的特征性匿名区域[10]。据研究报道Ψ区的缺失不会对狂犬病病毒的扩散和致病性等生命活动造成影响,同时也证实该区域有在神经细胞定位和表达外源基因的能力[2]。本研究通过缺失Ψ区进行病毒的拯救及鉴定,为后续的研究工作提供了理论依据。
狂犬病病毒为单股负链RNA病毒,在复制表达的过程中不需要DNA阶段,只在构建狂犬病病毒基因组感染性cDNA时,对其进行分子水平的操作[11]。裸露的狂犬病病毒基因组RNA无感染性,感染性狂犬病病毒的必须与N蛋白形成复合物,以保护RNA自身不被降解,同时还需要L蛋白与P蛋白配合充当RNA聚合酶的功能,共同构成具有活性的RNA复合物后,才可进行狂犬病病毒的转录和复制[10]。因此,狂犬病病毒的反向遗传操作系统,不仅需要病毒的全长基因组的参与,同时也需要N、P和L蛋白的协同。虽然G蛋白不是病毒拯救过程中必要组成成分,但已有相关研究认定若增加G蛋白可不同程度的提高狂犬病病毒的拯救效率[12-13]。狂犬病病毒的M蛋白可能对病毒的转录和复制起到调节作用,调节的程度主要与M蛋白浓度有关[14]。在狂犬病病毒研究过程中,大量文献主要是以添加3或4个辅助质粒(N、P和L蛋白或G蛋白)进行病毒的拯救[10,15-17],而本研究在建立狂犬病病毒SRV9 Ψ区缺失株反向遗传操作系统的电穿孔转染细胞部分的试验中不仅加入病毒全长感染性cDNA,同时加入了N、P、M、G和L各自的辅助表达质粒,提高了病毒的复制水平[18],进一步提高狂犬病病毒的拯救效率。
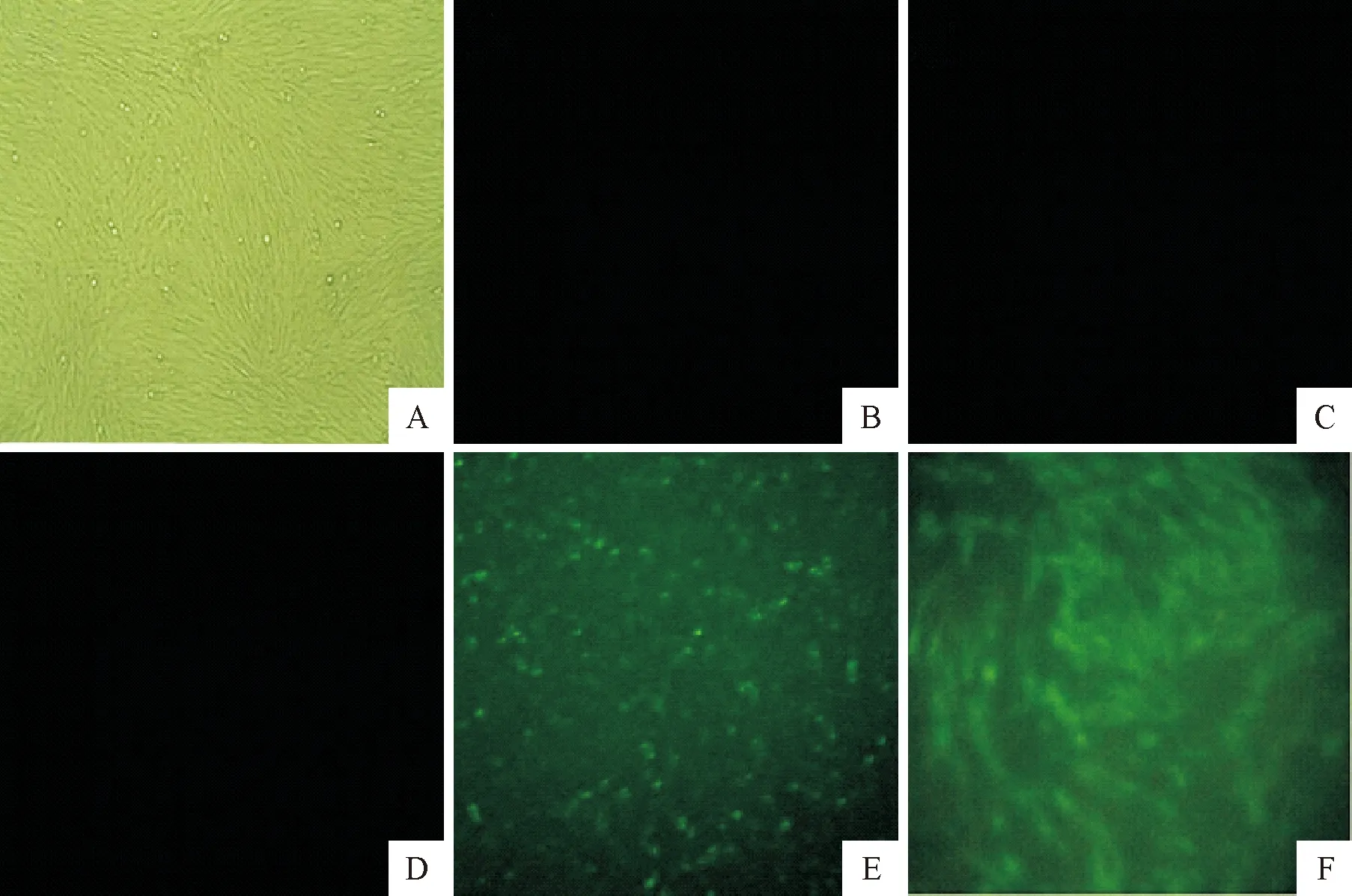
A.光学显微镜下正常BHK-21细胞;B.正常BHK-21细胞的阴性对照;C.只转染了辅助质粒;D.转染全长基因组表达质粒;E.重组病毒感染BHK-21;F.亲本毒株感染BHK-21阳性对照
A.Normal BHK-21 cells under light microscope; B:normal BHK-21 cells as negative control;C.Only transfection of auxiliary plasmid; D.Transfection of entire length plasmid; E.Recombinant virus infected BHK-21; F.Parent strain infected BHK-21 as positive control
图3间接免疫荧光检测结果(200×)
Fig.3 Indirect immunofluorescence test results(200×)

1.空白细胞;2.亲本毒株病毒液;3.重组毒株病毒液
1.Blank cells; 2.Recombinant virus; 3.Parent strain
图4 Western-blot检测G蛋白水平结果
Fig.4 Detection of G protein levels by Western-blot
Schnell M J等[2]首次成功建立了狂犬病病毒(SADB19株)的反向遗传操作系统。随后,Inoue K等[19]在病毒全长基因组感染性cDNA的3′端添加HamRz、5′端添加HdvRz序列。将HEP-Flury株的病毒cDNA插入至真核表达载体pcDNA3.1的CMV启动子下游,利用真核细胞聚合酶Ⅱ启动cMV启动子的高效表达,在不同的细胞系上拯救出HEP-Flury株。本研究将狂犬病病毒SRV9 Ψ区缺失株感染性cDNA连接真核表达载体pcDNA3.1的CMV启动子下游,同时在基因组的两端分别添HamRz和HdvRz,利用BHK-21细胞,在细胞体内完成重组DNA反转录,成功拯救出具有感染性的缺失Ψ区狂犬病病毒株。
细胞转染方法有脂质体法、电穿孔法、磷酸钙沉淀法及逆转录病毒介导转染法[20-21]。电穿孔转染细胞法有效的将外源基因导入哺乳动物细胞内,操作简单、安全性好、转染效率高适用性较为广泛,但由于较大的电压刺激使得部分细胞经电转后死亡[5]。通过多次的电穿孔转染细胞法总结发现:①转染时需使用最佳的细胞状态,转染成功率会增加。②转染时所用质粒一定是去除内毒素的高纯质粒,使用时进行分装,尽量避免反复冻融。③转染后拯救的新病毒不稳定,不要直接冻融收毒,在扩大培养五代以上后,待病毒稳定后,再进行后续试验。采用此方法能有效地提高大片段载体转染效率,本研究利用电穿孔转染细胞法进行病毒拯救,有效地提高了细胞转染效率。
本研究采用间接免疫荧光、RT-PCR、Western-blot和透射电子显微镜进行拯救毒株的鉴定,结果证实利用电穿孔转染法成功拯救出狂犬病病毒SRV9 Ψ区缺失株,验证了缺失Ψ区的病毒株并不影响G蛋白的表达。通过对亲本毒株与拯救毒株电子显微镜的形态观察,结果发现缺失Ψ区后狂犬病病毒的形态仍保持原有弹状形态,说明缺失Ψ对狂犬病病毒的形态影响较小。
本研究成功建立了狂犬病病毒SRV9 Ψ区缺失株的反向遗传学操作系统,利用电转染法能够提高转染效率,为进一步研究狂犬病病毒SRV9株基因组的结构功能间的关系及后续开展狂犬病相关新型疫苗研究等奠定了基础。

A.亲本SRV9株的病毒液负染后透射电镜观察;B.重组病毒液负染后透射电镜观察
A.Negative staining parent strain SRV9 observed by electron microscope; B.Negative staining recombinant virus observed by electron microscope
图5透射电子显微镜观察结果
Fig.5 Transmission electron microscopy images

A.亲本SRV9株的病毒颗粒电镜观察;B.拯救病毒颗粒电镜观察
A.Viral particles of parent strain SRV9 observed by electron microscope; B.Rescued recombinant viral particles observed by electron microscope
图6透射电子显微镜观察结果
Fig.6 Transmission electron microscopy images
[1] 扈荣良,张守峰,刘 晔.我国狂犬病预防和控制建议[J].中国人兽共患病学报,2012,28(5):487-491.
[2] Schnell M J.Infectious rabies viruses from cloned cDNA.[J].Embo J,1994,13(13):4195-4203.
[3] 刘晓娟,赵晓飞,曾贝妮,等.电穿孔法转染293T细胞条件的优化[J].生物学通报,2014,49(11):52-54.
[4] 张禾璇,单可人,何 燕,等.脂质体法与电穿孔法转染两种细胞效率的比较[J].重庆医学,2014(33):4432-4433.
[5] Takei H,Baba Y,Hisatsune A,et al.Glycyrrhizin inhibits interleukin-8 production and nuclear factor-kappaB activity in lung epithelial cells,but not through glucocorticoid receptors.[J].J Pharmacol Sci,2008,106(3):460-468.
[6] 卢 莎,谭文杰,张 玲,等.脂质体转染法与电转染法在丙型肝炎病毒RNA 转染人肝癌细胞中的应用效果比较[J].山东医药,2016,56(19):15-18.
[7] 李 茂.动物狂犬病弱毒疫苗生产工艺的建立及免疫试验研究[D].吉林大学,2007.
[8] 侯世宽,岳军明.狂犬病口服疫苗株的筛选、鉴定和实验免疫研究[C]:全国狂犬病、布氏菌病、弓形虫实验诊断新技术讨论会,1995.
[9] 钱爱东,侯世宽.狂犬病无毒疫苗株SRV9G基因主要功能区的序列分析[J].中国预防兽医学报,1997(6):20-23.
[10] 薛向红.狂犬病病毒CVS-11株感染性cDNA克隆的构建及其应用[D].吉林吉林:吉林大学,2014.
[11] Conzelmann K K.Reverse genetics of mononegavirales:The rabies virus paradigm[M].Springer Japan,2013.
[12] Sato S,Ohara S,Tsutsui K,et al.Effects of G-gene deletion and replacement on rabies virus vector gene expression[J].PLoS One,2015,10(5):e128020.
[13] Morimoto K,Foley H D,Mcgettigan J P,et al.Reinvestigation of the role of the rabies virus glycoprotein in viral pathogenesis using a reverse genetics approach.[J].J Neurovirol,2009,6(5):373-381.
[14] Finke S,Conzelmann K K.Dissociation of rabies virus matrix protein functions in fegulation of viral RNA synthesis and virus assembly[J].J Virol,2003,77(22):12074-12082.
[15] Huang Y,Tang Q D S,Zhang S,et al.Development of a reverse genetics system for a human rabies virus vaccine strain employed in China[J].Virus Res,2010,149(1):28-35.
[16] Wu X,Rupprecht C E.Glycoprotein gene relocation in rabies virus[J].Virus Res,2008,131(1):95-99.
[17] 刘 澜,李士成,陈小云,等.重组表达3G蛋白狂犬病病毒株的构建及重组病毒相关特性的研究[J].中国兽医科学,2016(3):326-331.
[18] Mebatsion T,Weiland F,Conzelmann K K.Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G[J].J Virol,1999,73(1):242-250.
[19] Inoue K,Shoji Y,Kurane I,et al.An improved method for recovering rabies virus from cloned cDNA.[J].J Viro Meth,2003,107(2):229-236.
[20] 李 进,洪光祥,王发斌,等.Lipofectamine介导转染神经干细胞的研究[J].中华实验外科杂志,2005,22(3):357-358.
[21] 赵 宇,陆应麟,乔 群,等.绿色荧光蛋白基因逆转录病毒载体转染人骨髓间充质干细胞及表达的研究[J].中华实验外科杂志,2004,21(1):24-25.
RescueandIdentificationofRabiesVirusSRV9ΨAeraDelectionstrainbyElectroporationTransfection
MAO Li-ping,WEI Yu-yuan,WANG Wei,ZHAI Shao-hua,CHENG Yao,WEN Zhao-hai,JIAN Zi-jian
(CollegeofVeterinaryMedicine,XinjiangAgriculturalUniversity,Urumqi,Xinjiang,830052,China)
The aim of the study was to investigate the rescue and identification of recombinant virus of SRV9 without pseudo gene Ψ region via electroporation transfection.The purified deleted pseudo gene Ψ region pCDNA-NPMG-L and the helper plasmids pcDNA-N,pcDNA-P,pcDNA-M,pcDNA-G and pcDNA-L were co-transfected into BHK-21 cells via electroporation transfection for virus rescue,and the BHK21 cells were used for blind passages of the recombinant rabies virus.The recombinant rabies virus SRV9 deleted pseudo gene Ψ region was identified by RT-PCR,indirect immunofluorescence,Western-blot and electron microscopy.The results showed that the recombinant virus could produce green fluorescence in the indirect immunofluorescence assay,the typical rhabdovirus was observed by transmission electron microscopy.The recombinant strain and the parent strain G protein were subjected to Western-blot to obtain the corresponding target band.The above results showed that transfection through electroporation cells in this experiment was able to rescue its recombinant virus SRV9 ψ region deletion strain.This study provided a new transfection method for rescue of virus,and can effectively improve the transfection efficiency,and provide effective theoretical basis and practical basis for the development of new rabies virus vaccine.
Rabies virus; electroporation transfection; SRV9 deleted pseudo gene Ψ region; rescue
2017-04-02
国家自然科学基金项目(31360623);研究生科研创新项目(XJAUGRI2015017)
毛丽萍(1989-),女,安徽砀山人,硕士研究生,主要从事动物分子与免疫病理学研究。△同等贡献作者。
*
S852.659.6
A
1007-5038(2017)11-0022-06
