应用Gibson Assembly方法构建狂犬病病毒SRV9株重组感染性cDNA克隆
2017-12-15魏玉圆翟少华毛丽萍皮文辉简子健
王 伟,魏玉圆,翟少华,毛丽萍,皮文辉,简子健*
(1.新疆农业大学动物医学学院,新疆乌鲁木齐 830052; 2.新疆生产建设兵团绵羊遗传改良和健康养殖重点实验室,新疆石河子 832000)
应用Gibson Assembly方法构建狂犬病病毒SRV9株重组感染性cDNA克隆
王 伟1,魏玉圆1,翟少华1,毛丽萍1,皮文辉2,简子健1*
(1.新疆农业大学动物医学学院,新疆乌鲁木齐 830052; 2.新疆生产建设兵团绵羊遗传改良和健康养殖重点实验室,新疆石河子 832000)
应用Gibson Assembly 连接法精确快速构建狂犬病病毒SRV9株重组感染性cDNA克隆,为狂犬病病毒感染性cDNA克隆的构建提供新的方法。应用Gibson Assembly 连接法在同一反应体系内,将多个片段和经限制性内切酶线性化的载体,按设计的顺序进行连接,实现多个片段的一步组装。设计带有20 bp~30 bp重叠序列的PCR引物,扩增得到带有重叠序列的狂犬病病毒5个结构蛋白基因和增强型荧光蛋白基因。扩增的片段和线性化的载体经胶回收纯化后,与Gibson连接液混合后,50℃反应60 min,连接产物转化Stbl3感受态细胞,提取重组质粒,经PCR、酶切和测序鉴定,缺失了病毒伪基因Ψ区和糖蛋白基因的跨膜区并且将增强型荧光蛋白基因插入糖蛋白基因终止密码子前,构建了全长17 600 bp的重组质粒,完成了重组全长感染性cDNA克隆的构建。
狂犬病病毒;Gibson Assembly连接法;构建;感染性cDNA克隆
我国属于狂犬病的高发国家,研制新型、安全、稳定、高效的疫苗是有效控制狂犬病的重要手段。狂犬病病毒(Rabies virus,RABV)是第一个利用反向遗传学操作系统拯救的单股负链RNA病毒[1],全长感染性cDNA克隆的构建是病毒成功拯救的关键所在。因此,精确快速地构建RABV全长感染性cDNA克隆,对于开发和研制新型疫苗至关重要。Gibson Assembly连接法自2009年报道以后,被广泛的用于人工基因组的合成[2-3],表达载体的构建[4],病毒感染性cDNA克隆的构建[5-6],CRISPR/Cas9基因编辑系统的组装[7],极大地促进了基因编辑技术的发展。
RABV为单股负链不分节段的RNA病毒,基因组全长为12 kb左右,包含5个结构蛋白基因:核蛋白(nucleoprotein,N)基因全长1 423 bp,编码450个氨基酸;磷蛋白(phosphoprotein,P)基因全长990 bp,编码297个氨基酸;基质蛋白(matrix protein,M)基因全长803 bp,编码202个氨基酸;糖蛋白(glycoprotein,G)基因全长2 069 bp,编码524个氨基酸;RNA依赖的RNA聚合酶(RNA dependent RNA polymerase,L)基因全长6 384 bp,编码2 127个氨基酸。将病毒全长cDNA插入真核表达载体CMV启动子下游,利用真核细胞聚合酶Ⅱ驱动CMV起始转录,外加辅助包装蛋白,从而启动病毒基因组的转录和复制,能够组装出完整的病毒颗粒[8]。全长真核表达质粒即感染性cDNA克隆的构建,是建立RABV反向遗传学操作系统的关键。在RABV反向遗传学操作系统的基础上,利用基因编辑技术实现定点突变、敲除、插入等,拯救出改造后的病毒可为研制新型、安全、稳定、高效的疫苗提供基础[9]。将标记基因eGFP定向插入特定位点,实现对RABV的标记,有利于研究RABV的致病机制和中和抗体快速测定[5,10-11]。精确快速的组装RABV全长重组质粒,对RABV新型疫苗的研制、致病机制的研究、病毒的开发利用和基因功能的研究具有重要意义。
本研究采用Gibson Assembly法将糖蛋白基因(G基因)由原来的第4位移至第2位,缺失了G基因的CD区132个碱基和伪基因Ψ区的423个碱基。在G基因的终止密码子前定点插入了eGFP增强型荧光蛋白基因。成功组装了狂犬病病毒SRV9株重组全长感染性cDNA克隆,实现了基因片段的精确快速组装,为RABV反向遗传系统的建立、研究提供基础。
1 材料与方法
1.1 材料
pcDNA3.1+载体,Stbl3菌种为Invitrogen公司产品,pMD19-T-N,pMD19-T-G,pMD19-T-PM,pMD19-T-L1,pMD19-T-L2,pMD19-T-L3均由新疆农业大学病理实验室构建。Gibson Assembly组装预混液为NEB公司产品,DL 2 000 DNA Marker,Primer STAR DNA Polymerase为日本TaKaRa公司产品;胶回收试剂盒,小量质粒提取试剂盒为美国OMEGA公司产品;限制性内切酶为美国Thermo Fisher公司产品; peGFP-C1质粒为北京鼎国昌盛生物技术有限责任公司产品。
1.2 方法
1.2.1 引物的设计 根据GenBank中发表的SRV9(AF499686)和peGFP-C1的eGFP基因序列设计Gibson连接引物,为方便后续检测设计了2对检测引物(表1)。通过锤头状核酶和丁型肝炎核酶的切割作用产生精确地3′末端和5′末端可提高病毒的拯救效率[12]。本研究采用PCR的方法将核酶序列添加到了全长质粒的两端,合成序列为:
F:TGTTAAGCGTCTGATGAGTCCGTGA- GGACGAAACTATAGGAAAGGAATTCCTAT- AGTCACGCTTAACAACCAGATCAAAG(下划线为锤头状核酶序列);
R:GGTCGGCATGGCATCTCCACCTCCTC- GCGGTCCGACCTGGGCATCCGAAGGAGGAC- GCACGTCCACTCGGATGGCTAAGGGAGGGC- GGGTACCACGCTTAACAAATAAACAAC(下划线为丁型肝炎核酶序列)。运用上游引物F和下游引物NR扩增N基因,上游引物L3F和下游引物R扩增L3基因,可将核酶序列添加到全基因组的两端。本研究在构建重组质粒pMD19-T-N和pMD19-T-L3时,将核酶序列通过PCR加入。

表1 引物名称及序列
1.2.2 狂犬病病毒结构蛋白基因和eGFP基因的扩增 以pMD19-T-N、pMD19-T-G、pMD19-T-PM、pMD19-T-L1、pMD19-T-L2、pMD19-T-L3、peGFP-C1质粒为模板PCR扩增狂犬病病毒SRV9株各结构蛋白基因和eGFP增强型荧光蛋白基因。L基因全长为6 475 bp,分为L1、L2、L3三段扩增。PM基因中间没有调整,所以可以一起扩增。
PCR反应体系:PrimeSTAR Max mix(2×)25 μL;forward primer 1 μL;reverse primer 1 μL;模板 0.5 μL;RNase Free dH2O 22.5 μL。PCR反应条件: 98℃,10 s,60℃,15 s,72℃,15 s,35个循环;72℃ 5 min。PCR产物于10 g/L 琼脂糖凝胶中电泳。按照OMEGA 高纯度DNA胶回收纯化试剂盒说明书进行操作。
1.2.3 重组质粒pcDNA-L的构建 用NheⅠ限制性内切酶 (Takara公司)过夜酶切pcDNA3.1+质粒,50 μL反应体系:10 mol/L buffer 5 μL、质粒5 μg、NheⅠ 1 μL、补水至50 μL。酶切产物用OMEGA胶回收试剂盒回收纯化。
按照NEB公司Gibson Assembly Master Mix试剂盒操作说明,组装片段摩尔数是载体的3倍,配置10 μL连接体系:NheⅠ酶线性化的pcDNA3.1+0.035 4 pmols,L1、L2、L3均为0.106 2 pmols,补水至5 μL,Gibson Assembly Master Mix(2×)连接液5 μL,混匀,PCR仪控温50℃反应60 min,反应结束后立即放在冰上进行转化。
将连接产物加入100 μL Stbl3感受态细胞中,冰浴30 min,42℃水浴1 min,立即放在冰上2 min~3 min,加入800 μL LB培养基200 r/min、37℃培养1 h,3 500 r/min离心10 min,弃上清800 μL,轻轻的吹打混匀后涂布于含Amp的平板上,37℃培养12 h~14 h。
1.2.4 pcDNA-L重组质粒的菌体PCR检测 挑取转化的单菌落在含Amp的平板上划线,37℃培养8 h,挑取少量菌体利用设计的检测引物JCLF/JCLR进行PCR扩增。配制25 μL反应体系:10×Taqbuffer 2.5 μL;dNTP Mix(2.5 mmol/L),2 μL;上下游引物各1 μL;TaqDNA polymerase 0.25 μL; RNase Free dH2O,18.25 μL。PCR反应条件:94℃ 3 min;94℃ 30 s,60℃ 30 s,72℃ 60 s,35个循环;72℃ 10 min;4℃终止反应。取5 μL PCR产物于10 g/L琼脂糖凝胶中电泳。
1.2.5 pcDNA-L重组质粒的酶切鉴定 用BamHⅠ酶切重组质粒产生187、2 361、2 869、6 666 bp大小的4个片段。酶切体系Fast Digest buffer(10×)2 μL;pcDNA-L、500 ng;BamHⅠ 1 μL;补水至15 μL。37℃水浴2 h,8 g/L的琼脂糖凝胶电泳检测。
1.2.6 重组质粒pcDNA-N-G-eGFP-PM-L的构建 全长重组质粒构建示意图(图1)。将鉴定正确的1.2.5中的重组质粒pcDNA-L,用NheⅠ限制性内切酶线性化后,胶回收纯化,按照NEB公司Gibson Assembly Master Mix试剂盒操作说明配置10 μL连接体系:线性化的pcDNA-L质粒0.047pmols,N、G、PM、eGFP均为0.141 pmols, 补水至5 μL,Gibson Assembly Master Mix(2×)5 μL轻轻混匀,PCR仪控温50℃反应60 min,反应结束后立即放在冰上进行转化。转化步骤同1.2.3中转化步骤。菌体PCR引物为JCNGF/JCNGR,体系同1.2.4中菌体PCR检测体系。
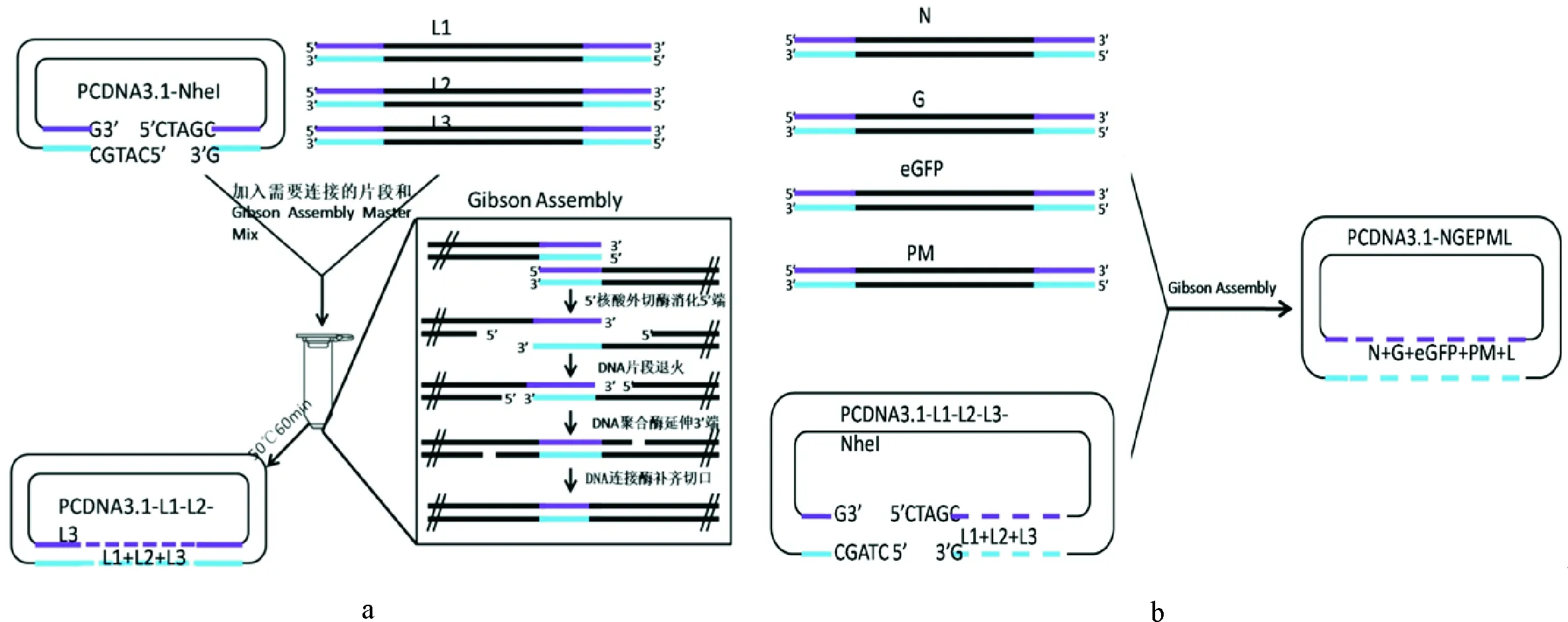
a.重组质粒pcDNA-L的组装;b.重组质粒pcDNA-N-G-eGFP-PM-L的组装a.Recombinant plasmid pcDNA-L assembly; b.Recombinant plasmid pcDNA-N-G-EGFP-PM-L assembly
1.2.7 pcDNA-N-G-eGFP-PM-L重组质粒的酶切鉴定 用BamHⅠ酶切重组质粒会产生187、930、1 844、2 361、2 869、9 409 bp大小的6个片段。酶切体系Fast Digest buffer(10×)、2 μL;重组质粒、1 μg;BamHⅠ、1 μL;补水至15 μL。37℃水浴2 h,8 g/L的琼脂糖凝胶电泳检测。XhoⅠ酶切会产生3 842、5 705、7 408 bp 3个片段酶切体系与BamHⅠ酶切体系相同。取经过菌体PCR检测为阳性,酶切鉴定正确的重组质粒50 μL,送上海生工生物工程技术服务有限公司测序。
2 结果
2.1 狂犬病病毒SRV9株各蛋白基因和eGFP基因的PCR扩增
以各蛋白基因重组质粒为模板,用设计的Gibson连接引物扩增各基因片段,用于Gibson连接的引物含有重叠片段,所以引物比较长。PCR扩增得到N基因1 589 bp、G基因1 473 bp、PM基因1 849 bp、eGFP基因763 bp、L1基因2 116 bp、L2基因2 272 bp、L3基因2 194 bp。结果显示每个基因片段扩增正确(图2),5′端的修饰基本不影响扩增效果,经胶回收纯化后可直接用于Gibson连接。

M1.DNA 标准 DL 2 000; 1.N; 3.G; 5.PM; 7.L1; 9.L2; 11.L3; 13.eGFP; 2、4、6、8、10、12.阴性对照; M2.DNA标准 DL15 000
M1.DNA Maker DL 2 000; 1.N; 3.G; 5.PM; 7.L1; 9.L2; 11.L3; 13.eGFP; 2,4,6,8,10,12.Negative control; M2.DNA Maker DL15 000
图2 PCR扩增结果
Fig.2 The result of PCR amplification
2.2 全长重组质粒的菌体PCR检测
分别用设计的检测引物JCNGF/JCNGR扩增序列跨N基因和G基因,按设计顺序连接理论片段大小为889 bp(图3a);JCLF/JCLR菌体PCR检测理论片段大小为719 bp(图3b)。菌体PCR初步可以判断是按照设计顺序连接成功。
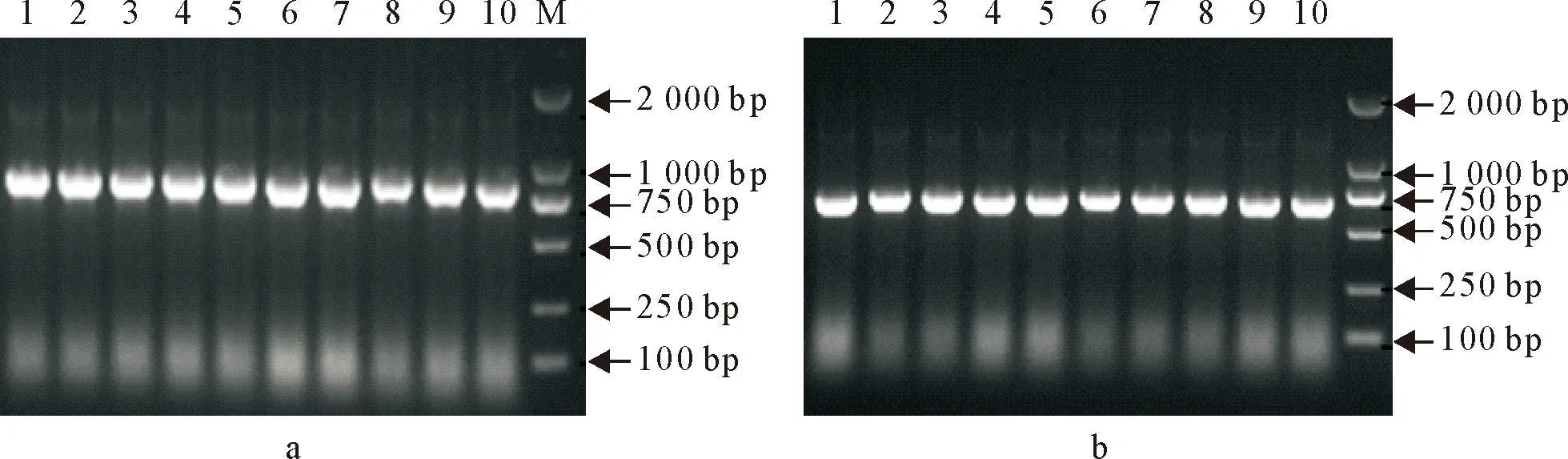
a 1~10.引物 JCLF/JCLR 菌体PCR结果; M.DNA Marker DL 2 000;b 1~10.引物JCNGF/JCNGR菌体PCR 结果; M.DNA Marker DL 2 000
a 1-10.Primer JCLF/JCLR bacteria PCR results; M.DNA Marker DL 2 000; b 1-10.Primer JCNGF/JCNGR bacteria PCR results; M.DNA Marker DL 2 000
图3菌体PCR电泳检测
Fig.3 The result of bacteria PCR detection
2.3 重组质粒的酶切鉴定
选择合适的限制性内切酶酶切重组质粒,如果电泳呈现的DNA片段大小与预期的片段大小一致,说明重组质粒是按照设计的顺序连接。pcDNA-L用BamHⅠ酶切重组质粒,预期产生187、2 361、2 869、6 666 bp大小的4个片段,XhoⅠ酶切产生3 842 bp、8 241 bp两个片段(图4a)。pcDNA-N-G-eGFP-PM-L全长重组质粒用BamHⅠ酶切重组质粒,预期产生187、930、1 844 、2 361、2 869、9 409 bp大小的6个片段。XhoⅠ酶切产生3 842、5 705、7 408 bp 3个片段(图4b)。电泳结果显示全长重组质粒pcDNA3.1-N-G-eGFP-PM-L酶切片段大小与预期结果一致。
2.3 全长质粒的测序结果
测序结果显示,G基因由原来的第四位M基因后移至第2位N基因后,敲除了G基因的跨膜区(CD区),并在其终止密码子前插入了eGFP增强型荧光蛋白标记基因。完全按照预期试验设计进行组装,连接部位完全正确。全长测序结果与NCBI公布结果进行比对,L基因由三处碱基突变。可能原因为PCR扩增过程中的错配(图5)。
(a) M1.DNA标准DL15 000; 1.plasmid pcDNA-L;2.XhoⅠ; 3.BamHⅠ;M2.DNA标准 DL 2 000;(b) M1.DNA 标准 DL15 000; 1.plasmid pcDNA-N-G-eGFP-PM-L;2.XhoⅠ;3.BamHⅠ;M2.DNA 标准DL 2 000
(a) M1.DNA Maker DL15 000; 1.plasmid pcDNA-L;2.XhoⅠ; 3.BamHⅠ;M2.DNA Maker DL 2 000;(b) M1.DNA Maker DL15 000; 1.plasmid pcDNA-NGEPML;2.XhoⅠ; 3.BamHⅠ;M2.DNA Maker DL 2 000
图4重组质粒酶切pcDNA-L(a)和pcDNA-N-G-eGFP-PM-L(b)
Fig.4 Restriction enzyme digestion of recombinant plasmid PCDNA-L(a)and PCDNA-N-G-EGFP-PM-L(b)
图5 重组质粒pcDNA-N-G-eGFP-PM-L测序结果
3 讨论
目前RABV感染性cDNA克隆的构建多采用传统的酶切连接的方法,此方法需要选择合适的酶切位点,将多个片段逐个连接[5,10-11]。利用此方法进行基因插入时,会在连接片段之间残存限制性内切酶的结合位点,无法实现基因的精确编辑。对于基因的定点突变、敲除就更加困难。Gibson Assembly连接法构建RABV感染性cDNA克隆时,利用片段之间的重叠序列,不额外的添加任何碱基,并且一次可连接多个片段,大大提高了连接效率。
ⅡS型限制性内切酶(ⅡS restriction enzymes)的发现让多片段快速无缝连接成为可能。ⅡS型限制性内切酶的识别位点在切割位点之外,通过与传统的连接酶结合使用,实现了PCR产物的环化和基因片段的无缝连接[13-14]。Engler C等[15]利用ⅡS型限制性内切酶,成功实现了多个片段的无缝快速组装。采用“Goden gate”克隆法一步连接3个片段,成功构建了同源重组靶向载体[16]。利用此方法进行多个大片段连接时,受到 IIS 型限制性内切酶的识别位点限制,可能无法找到合适的IIS型限制性内切酶。因此,无法用于RABV感染性cDNA的构建。
Gibson D G等[3]发明的Gibson Assembly连接法,利用DNA片段之间的重叠序列,实现了一步将多个DNA片段按特定顺序“无缝”连接。此方法利用T5 核酸外切酶(T5 exonuclease)、Phusion DNA polymerase、TaqDNA Ligase 3个酶在50℃时都有活性,利用基因片段之间的一小段重叠序列将多个片段连接起来。利用此方法,设计60 bp的重叠序列,成功构建了16.3 kb的小鼠线粒体基因组[17]。这一技术还被用来构建了第一个人造细胞的基因组[2]。戢志呈等[4]利用Gibson Assembly连接法设计20 bp~25 bp互补重叠序列成功快速的构建了多个植物表达载体。Gibson Assembly连接法连接片段的长度可以是几百个碱基,最长可以达到上百kb[3]。目前Gibson Assembly连接法已经被用于CRISPR/Cas9系统的构建,病毒感染性cDNA克隆的构建等[6-7,18]。因此该方法适用于各种载体的构建,可实现DNA片段的“无缝”连接、基因敲除、插入。
在构建狂犬病病毒感染性cDNA克隆时,为了产生精确末端,多会在全长基因组的两端引入锤头状核酶和丁型肝炎核酶序列,研究者们多将核酶序列利用基因合成技术合成后,利用酶切连接成功实现了核酶序列的插入[9-10]。本试验直接将核酶序列合成到PCR扩增的引物中,实现了核酶序列的插入。虽然引物序列较长,试验结果表明,基本不影响基因片段的扩增。
利用Gibson Assembly连接法进行狂犬病病毒感染性cDNA克隆的构建有如下优点:①可以一步实现多个不同大小片段按照设计顺序组装,方便快捷。②可以实现基因的精确插入,本研究将eGFP基因插入G基因终止密码子前,实现了基因片段的定点插入。③可以实现基因敲除,本研究成功敲除了G基因CD区的132个碱基,编码43个氨基酸。④实现了基因片段的“无缝”连接,在连接片段之间不额外的添加碱基,有利于RABV基因功能的研究。
本研究采用Gibson Assembly连接法将G基因由原来的第4位移至第2位,说明此方法可以将RABV的5个结构蛋白基因按照设计的顺序进行组装。eGFP标记基因的插入,说明利用Gibson连接法可以精确的插入外源基因。对Ψ区和CD区的敲除说明Gibson Assembly连接法可以实现任何位置的基因敲除。利用Gibson Assembly连接法可以快速构建各种表达载体。
[1] Schnell M J,Mebatsion T,Conzelmann K.Infectious rabies viruses from cloned cDNA.[J].EMBO J,1994,13(18):4195.
[2] Gibson D G,Glass J I,Lartigue C,et al.Creation of a bacterial cell controlled by a chemically synthesized genome[J].Science,2010,329(5987):52-56.
[3] Gibson D G,Young L,Chuang R,et al.Enzymatic assembly of DNA molecules up to several hundred kilobases[J].Nat Meth,2009,6(5):343-345.
[4] 戢志呈,江雁翔,刘耀光,等.应用Gibson Assembly方法构建植物表达载体[J].华南农业大学学报,2014(5):112-116.
[5] Tang H,Lu Z,Wei X,et al.A recombinant rabies virus expressing a phosphoprotein-eGFP fusion is rescued and applied to the rapid virus neutralization antibody assay[J].J Virol Meth,2015,219:75-83.
[6] Blawid R,Nagata T.Construction of an infectious clone of a plant RNA virus in a binary vector using one-step Gibson Assembly[J].J Virol Meth,2015,222:11-15.
[7] Wang J,Wang A,Li K,et al.CRISPR/Cas9 nuclease cleavage combined with Gibson assembly for seamless cloning[J].Biotechniques,2015,58:161-170.
[8] Inoue K,Shoji Y,Kurane I,et al.An improved method for recovering rabies virus from cloned cDNA[J].J Virol Meth,2003,107(2):229-236.
[9] 解庭波,明平刚,唐 芳,等.狂犬病病毒CTN株反向遗传系统的改造及拯救[J].中国生物制品学杂志,2015,28(11):1121-1126.
[10] 薛向红,郑学星,盖微微,等.狂犬病病毒CVS-11株全序列测定及其感染性克隆的构建[J].微生物学报,2013,53(4):409-415.
[11] Wickersham I R,Finke S,Conzelmann K,et al.Retrograde neuronal tracing with a deletion-mutant rabies virus[J].Nat Meth,2007,4(1):47-49.
[12] Takayama-Ito M,Inoue K I,Shoji Y,et al.A highly attenuated rabies virus HEP-Flury strain reverts to virulent by single amino acid substitution to arginine at position 333 in glycoprotein[J].Virus Res,2006,119(2):208-215.
[13] Kotera I,Nagai T.A high-throughput and single-tube recombination of crude PCR products using a DNA polymerase inhibitor and type IIS restriction enzyme[J].J Biotechnol,2008,137(1-4):1-7.
[14] Engler C,Kandzia R,Marillonnet S.A one pot,one step,precision cloning method with high throughput capability[J].PLoS One,2008,3(11):e3647.
[15] Engler C,Gruetzner R,Kandzia R,et al.Golden gate shuffling:a one-pot DNA shuffling method based on type IIs restriction enzymes[J].PLoS One,2009,4(5):e5553.
[16] 梁 龙,杨 华,杨永林,等.“Golden Gate”克隆法构建靶向载体[J].中国生物工程杂志,2013(3):111-116.
[17] Gibson D G,Smith H O,Hutchison III C A,et al.Chemical synthesis of the mouse mitochondrial genome[J].Nat Meth,2010,7(11):901-903.
[18] Bordat A,Houvenaghel M,German-Retana S.Gibson assembly:an easy way to clone potyviral full-length infectious cDNA clones expressing an ectopic VPg[J].Virol J,2015,12(1):1.
ConstructionofRecombinantInfectiouscDNACloneofSRV9RabiesVirusbyGibsonAssembly
WANG Wei1,WEI Yu-yuan1,ZHAI Shao-hua1,MAO Li-ping1,PI Wen-hui2,JIAN Zi-jian1
(1.CollegeofVeterinaryMedicine,XinjiangAgriculturalUniversity,Urumqi,Xinjiang,830052,China; 2.StateKeyLaboratoryforSheepGeneticImprovementandHealthyProduction,XinjiangAcademyofAgriculturalandReclamationSciences,Shihezi,Xinjiang,832000,China)
The recombinant full-length infectious cDNA clone of SRV9 rabies virus was constructed by Gibson Assembly,which provided a new method for the construction of rabies virus infectious cDNA clone.A few fragments and the enzyme linearized carriers were connected by Assembly Gibson connection method in the same reaction system according to the design of connection order for the realization of one step assembly of a number of fragments.PCR primers were designed with 20-30bp overlap sequence,and five structural protein genes and fluorescent protein gene with overlapping sequences were amplified.The amplified fragments and linearized vectors after purification by gel extraction were mixed and connected to the Gibson liquid,reacting at 50℃ for 60 min,the products were transformed into Stbl3 competent cells.The recombinant plasmid was identified by PCR,enzyme digestion and sequencing.After the deletion of the glycoprotein gene transmembrane region and virus pseudo gene ψ region inserting enhanced green fluorescent protein gene into ahead the glycoprotein gene stop codon,the 17 600 bp recombinant plasmid and the recombinant infectious full length cDNA clone were constructed successfully and quickly.
Rabies virus; Gibson Assembly;construction;infectious cDNA clone
2017-03-19
国家自然科学基金项目(31360623);新疆生产建设兵团应用基础研究项目(2016AG008)
王 伟(1990-),男,新疆巩留人,硕士研究生,主要从事动物分子与免疫病理学研究。*
S852.659.1
A
1007-5038(2017)11-0011-07
