新型4-羟基香豆素衍生物的合成与表征
2017-11-30权彦李小蓉赵忠孝刘靖丽张朋
权彦,李小蓉,赵忠孝,刘靖丽,张朋
新型4-羟基香豆素衍生物的合成与表征
权彦,李小蓉,赵忠孝,刘靖丽,张朋
(陕西中医药大学, 陕西 咸阳 712046)
香豆素是自然界天然产物中一类十分重要的有机化合物。以4-羟基香豆素为原料,采用微波合成法“一锅煮”法,合成了系列新型香豆素衍生物,所有产物进行了IR、MS、1HNMR结构表征,其中部分产物为首次合成。为此类抗凝血药物的研究提供了理论技术支持。
微波合成;香豆素;抗凝血;结构表征
4-羟基香豆素及其衍生物是一类较特殊的香豆素化合物,具有抗凝血,抗真菌、抗癌等多种生物生理学活性,多年来一直受到医药学家的高度重视[1]。香豆素的生物学活性因其结构而定,如在母体苯环或吡喃环引入不同的取代基或引入的位置不同而显示不同的生物活性[2]。开展对各种含不同取代基的香豆素类化合物的合成研究,并进一步研究其生物学活性具有重要的医学和生物学意义。
本文采用微波合成方法“一锅煮”法[3],将香豆素溶解,然后根据要合成的目标产物的结构,进行理论分析推导,选用含有相应基团结构的有机试剂;加入相应的原材料,探索加热温度,最后控制温度加热、搅拌、抽滤得目标化合物。合成技术路线如下:

1 实验部分
1.1 主要仪器与试剂
液体核磁共振仪 INOVA 500 MHz型 varian公司;BIO-RAD FTS3000型红外光谱仪(KBr压片) 202-1AB BRUKER VERTEX;质谱仪 6301安捷伦;熔点测定仪 RY-1型上海一恒科学仪器有限公司;电热真空干燥箱XRD-700上海一恒科学仪器有限公司;紫外可见分光光度计UV-2102尤尼科(上海)仪器有限公司;微波炉800W美迪集团;恒温加热搅拌仪781HW型常州电器有限公司。
乙醇(CH3CH2OH)天津市天力化学试剂有限公司;乙酸酐(CH3COOCOCH3)天津市天力化学试剂有限公司;4-羟基香豆素 西安光宇生物科技有限公司;3,4-二氯苯甲醛 兰州化学试剂厂;3,4-二甲基苯甲醛 庆功化学试剂有限公司;3-甲基苯甲醛 兰州化学试剂厂;4-甲基苯甲醛 庆功化学试剂有限公司;4-二甲氨基苯甲醛 南京有机试剂厂;以上试剂都为分析纯。去离子水 自制。
1.2 目标化合物的合成与结构表征
1.2.1 以化合物A进行试验对反应条件进行选择。
方案一:在250 mL三颈烧瓶中加入4-羟基香豆素10.0 g和无水乙醇100 mL,加热、搅拌至4-羟基香豆素溶解后,然后慢慢均匀从三颈烧瓶侧口再加入3,4-二甲基苯甲醛8 mL,固定好温度计,加热,温度控制在不同温度(如表1所示),回流4 h,后有白色固体颗粒析出,继续加热,温度不变,加热1 h,待反应结束,自然冷却后抽滤,得白色晶体,此晶体再用95%乙醇进行重结晶,最终得纯白色颗粒状晶体[4]。自然干燥后称量产物,计算产率。产品经自制蒸馏水重结晶2次后,干燥,测量化合物A熔点为322~323 ℃。
方案二:在250 mL三颈烧瓶中加入4-羟基香豆素10.0 g和无水乙醇100 mL,搅拌,然后慢慢均匀从三颈烧瓶侧口再加入3,4-二甲基苯甲醛8 mL,采用固定功率的微波炉加热,搅拌、回流冷却,加热不同时间(如表2所示),后有白色固体颗粒析出,继续微波加热6 min,待反应结束,自然冷却后抽滤,得白色固体,此固体再用95%乙醇进行重结晶,最终得纯白色粉末[5,6]。自然干燥后称量产物,计算产率。产品经自制蒸馏水重结晶2次后,干燥,测量化合物A熔点为322~323 ℃。
采用不同温度和800 W微波炉加热合成目标产物,探讨了反应温度对合成目标产物产率的影响,不同温度和微波加热不同时间时产率如表1和表2所示。

表1 反应温度对产率的影响
由表1可知,反应在85 ℃时产率最高,当温度超过100 ℃产率已经很低,且反应结束后三颈烧瓶中颜色明显变黑,这是由于高温时部分有机物碳化,最终分离提取时大部分都为杂质被除去,故产率明显降低,当温度低于80 ℃时,在分离提取时,大部分由于原料未反应而被除去,故产率明显降低。当温度在85 ℃时,产率达到最大值,故本合成反应温度控制在85 ℃。
由表2可知微波加热12 min或17 min产率达到43%,当微波加热23 min时,三颈瓶中有黑烟冒出,说明温度太高,大量物质碳化,最终产率0.3%,几乎无目标产物。加热时间低于10 min时,产率降低。
结合表1和表2分析可知,当通常情况水浴加热温度达到85 ℃时,加热时间4 h或微波加热15 min,目标产物的产率基本都达到最大值,故本实验合成目标产物A、B、C、D、E、F、G时采用此条件。
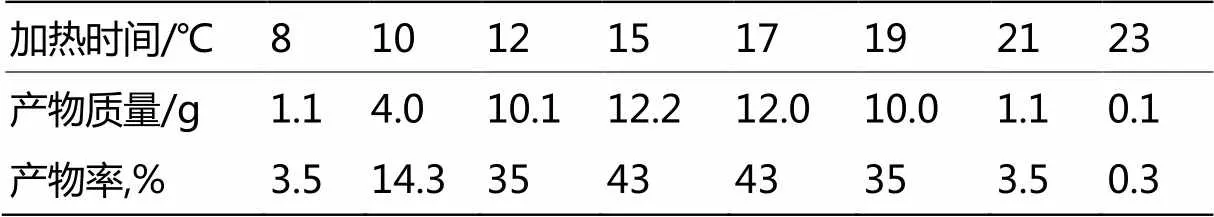
表2 微波加热不同时间对产率的影响
1.2.2 化合物1~7的合成
化合物1~7的合成参照相关化学文献方法合成。
化合物A的合成:在250 mL三颈烧瓶中加入4-羟基香豆素10.0 g和无水乙醇100 mL,加热、搅拌至4-羟基香豆素溶解后,然后慢慢均匀从三颈烧瓶侧口再加入3,4-二甲基苯甲醛8 mL,固定好温度计,加热,温度控制在85 ℃,回流4 h(或微波加热15 min)后有白色固体颗粒析出,继续加热,温度仍控制在85 ℃,加热1 h左右(或微波加热6分钟),待反应结束,自然冷却后抽滤,得白色固体,此晶体再用95%乙醇进行重结晶,最终得纯白色颗粒状固体[7]。自然干燥后称量产物12.1 g,产率43%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点322~323 ℃。
化合物B的合成:在250 mL三颈烧瓶中加入准确称取的4-羟基香豆素10.0 g和无水乙醇100 mL,加热、搅拌至4-羟基香豆素溶解后,然后慢慢均匀从三颈烧瓶侧口再加入3-甲基苯甲醛7 mL,固定好温度计,其他步骤同化合物A的合成步骤相同,最终得乳白色粉末。过夜,自然干燥后称量产物10.0 g,产率33%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点237~238 ℃。
化合物C的合成:合成方法同上,在已经溶解的4-羟基香豆素中慢慢加入4-甲基苯甲醛,重结晶,过滤,提纯,最终得乳白色固体15.0 g,计算产率为56%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点291~292 ℃。
化合物D的合成:在250 mL三颈烧瓶中加入4-羟基香豆素10.0 g和无水乙醇100 mL,加热、后加入KOH 10 mg,搅拌至4-羟基香豆素溶解后,然后慢慢均匀从三颈烧瓶侧口再加入4-二甲氨基苯甲醛,固定好温度计,加热,温度控制在80 ℃,回流4 h(或微波加热15 min)后有灰色固体粉末析出,继续加热,温度仍控制在80 ℃,加热1 h左右(或微波加热6 min),TLC检测反应反应结束后,自然冷却,蒸掉多余的乙醇,用乙酸乙酯萃取分离,多次萃取分离后抽滤,得灰色固体粉末,再用95%乙醇进行重结晶,最终得灰色固体[8,9]。自然干燥后称量产物12.0 g,产率41%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点119~220 ℃。
化合物E的合成:合成方法同化合物D相同,将原材料改为4-甲基苯甲醛,最终的白色粉末10克,产率36%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点269~270 ℃。
化合物F的合成:合成方法同A的合成相同,将原材料改为4-硝基苯甲醛,产品在自然干燥后称量产物10.0 g,产率35%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点251~252 ℃。
化合物G的合成:合成方法同A,将原材料改为3,4-二氯苯甲醛,最终产物自然干燥后称量产物13.0 g,产率43%。产品经自制蒸馏水重结晶2次后,干燥,测量熔点285~286 ℃。
1.2.3化合物8~12的合成
按照scheme1方案,将上述各步合成的A、B、C、D、E、F、G产物分别作为中间体,进行分子内脱水[10,11]。将相应的中间体化合物溶于乙酸酐中加热,搅拌,插入温度计,温度控制在90 ℃,回流4小时(或微波加热15 min),TLC监测反应,待反应结束后,自然冷却,分离萃取,抽滤,再用无水乙醇重结晶,最终分别得纯白、乳白、乳白、土灰色色固体8~12。
2 目标化合物的结构表征
对合成的7种化合物分别进行核磁共振(NMR)和质谱(MS)分析。利用熔点测定仪、红外光谱等仪器进行了测定,结果如下:
化合物A:
m.p.322-323°C.IR(KBr pelletcm-1): 3100, 2900, 1670, 1604, 1576, 1350, 1230,1093, 761 cm-1.1HNMR (CDCl3, δ, ppm): 11.507 (s,1H), 11.268(s, 1H),8.015–8.065(q,2H),7.606–7.647(t,2H),7.404–7.425(d, 4H), 7.067–7.088(d, 1H), 6.950(s, 2H), 6.049(s, 1H), 2.204–2.241(d, 6H). HRMS (ESI+):/: calcd for C27H20O6: 463.1152 [M+Na+]; found: 463.1139。
化合物B:
3,3′-(3-甲基苯亚甲基)-双-4-羟基香豆素(2): m.p. 237–238 °C. IR(KBr pellet cm-1): 1674, 1604, 1560, 1348, 1230,1101, 763 cm-1.1H NMR (CDCl3, δ, ppm): 11.528(s, 1H), 11.285(s, 1H), 8.009-8.088(q, 2H),7.623-7.654(t,2H),7.415-7.432(d,4H),7.206-7.236(t,1H),7.082-7.097 (d, 1H), 7.012-7.042 (t, 2H), 6.080 (s, 1H), 2.312(s,3H). HRMS (ESI+):/: calcd for C26H18O6: 449.0996 [M+Na+]; found: 449.0985。
化合物C:
3,3′-(4-甲基苯亚甲基)-双-4-羟基香豆素(3):m.p. 291–292 °C. IR(KBr pellet cm-1): 1670, 1618, 1564, 1352, 1230,1095, 906, 763 cm-1.1H NMR (CDCl3, δ, ppm): 11.521(s, 1H), 11.296(s, 1H), 8.000-8.082(q,2H),7.615-7.649(m, 2H), 7.408-7.425 (d, 4H), 7.099-7.145(q, 4H), 6.076(s, 1H), 2.342(s, 3H). HRMS(ESI+):/: calcd for C26H18O6: 449.0996 [M+Na+]; found: 449.0941。
化合物D:
3,3′-(4-二甲氨基苯亚甲基)-双-4-羟基香豆素(4): m.p. 119-220 °C. IR (KBr pellet cm-1): 1720, 1667, 1610, 1520, 1370, 1170, 756 cm-1.1HNMR (DMSO-6,, ppm): 7.788-7.803(d, 2H), 7.492-7.524(t, 2H), 7.390(s, 2H), 7.210-7.274(m, 6H), 6.267(s, 1H), 3.122(s, 6H). HRMS (ESI+):/: calcd for C27H21NO6: 478.1261 [M+Na+]; found: 478.1228。
化合物E:
3,3′-(4-甲氧基苯亚甲基)-双-4-羟基香豆素(5):m.p. 269-270 °C. IR(KBr pellet cm-1): 1670, 1604, 1560, 1510, 1350, 1256, 1093, 769 cm-1.1H NMR (CDCl3, δ, ppm): 11.512(s, 1H), 11.296(s, 1H), 7.994–8.075(q,2H),7.605–7.647(t, 2H), 7.399–7.419(d, 4H), 7.118–7.139(d, 2H), 6.844–6.866(d, 2H), 6.050(s, 1H), 3.797(s,3H). HRMS (ESI+):/: calcd for C26H18O7: 465.0945 [M+Na+]; found: 465.0967。
化合物F:
3,3′-(4-硝基苯亚甲基)-双-4-羟基香豆素(6): m.p. 251–252 °C. IR(KBr pellet cm-1): 1658, 1616, 1564, 1521, 1348, 1109, 763 cm-1.1H NMR (CDCl3, δ, ppm): 11.571(s, 1H), 11.379(s, 1H), 8.180–8.202(d, 2H), 7.997–8.104(q, 2H), 7.655–7.693(d, 2H), 7.404–7.443(t, 6H), 6.122(s, 1H). HRMS (ESI+):/: calcd for C25H15NO8: 480.0690 [M+Na+]; found: 480.0489。
化合物G:
3,3′-(3,4-二氯苯亚甲基)-双-4-羟基香豆素(7): m.p. 285-286 °C. IR(KBr pellet cm-1): 1670, 1604, 1560, 1350, 1101, 910, 767 cm-1.1H NMR (CDCl3, δ, ppm): 11.585(s, 1H), 11.317(s, 1H), 8.003–8.090(q, 2H), 7.639–7.678(t, 2H), 7.382–7.441(q, 5H), 7.285(s, 1H), 7.058–7.082(d, 1H), 6.021(s, 1H). HRMS (ESI+):/: calcd for C25H14Cl2O6: 503.0060 [M+Na+]; found: 503.0024。
3 结论
本实验按照实验方案,通过大量实验讨论了常压下加热至不同温度时合成的目标产物,同时采用800 W微波加热,测定了加热不同时的产率,最终得出结论,常压下加热4 h和微波加热15 h有着相同的产率,产率都达到最大值43%。故本实验合成时的温度控制在85 ℃,加热4 h或微波加热15 h。在合成化合物8~12时,用乙酸酐脱水,得到目标产物。
本文先对合成的7个香豆素衍生物进行熔点测定、红外、核磁等表征,推测出了它们的结构,结构推断7个化合物的结构分别为:

[1] Kostova I. Synthetic and natural coumarins as cytotoxic agents[J]. Cwr. Med Chem., 2005,5:29-46.
[2] 刘芳,兰支利,邓芳.香豆素及其衍生物研究进展[J].精细化工中间体,2006,36(4):7-11.
[3] 布文安.4,7一二羟基香豆素衍生物合成及杀菌活性研究[D].南京:南京农业大学,2008:3-22.
[4] 郭大鹏.香豆素衍生物的合成及活性检测[D].南京:南京理工大学,2008:1-23.
[5] 凌云, 杨旭, 杨明汉, 陈万义. 双香豆素类化合物的合成及表征. 化学通报[J], 2004, 67(5): 355-358.
[6] Li X, Jain N, Russell R K, Ma R, Branum S, Xu J, Sui Z. Development of a Scalable Synthetic Process for Selective Bromination of 4-Methyl-3,7-Substituted Coumarins [J]. Organic Process Research & Development, 2006, 10: 354-360.
[7] Li X, Jain N, Russfell R K, Ma R, Branum S, Xau J, Sui Z. Development of a Scalable Synthetic Process for Selective Bromination of 4-Methyl-3,7-Substituted Coumarins [J]. Organic Process Research & Development, 2006, 10: 355-370.
[8] 徐克勋. 精细有机化工原料及中间体手册[M]. 北京: 化学工业出版社, 2004: 4-54.
[9] 申东,刘小帆.香豆素类凝血药及其类似物的合成[J].应用化学,2005,22(10):1158-1160.
[10]严胜骄,林军.微波辅助1,4-迈克尔加成合成香豆素3,4-并六元杂环衍生物[J].有机化学,2010, 30(3): 465-468.
[11]凌云,杨旭,杨明汉,陈万义.双香豆素类化合物的合成及表征[J].化学通报, 2004,67(5): 355-358.
Synthesis and Characterization of Novel 4-Coumarin Derivatives
(Shaanxi University of Chinese Medicine, Shaanxi Xianyang 712046, China)
Coumarin is one of the most important organic compounds in natural products. In this paper, using 4- hydroxycoumarin as raw material, series of novel coumarin derivatives were synthesized by microwave synthesis method and one pot method, all products were characterized by IR, MS and1HNMR, some products were synthesized for the first time. The paper can provide theoretical and technical support for the study of anticoagulant drugs.
Microwave synthesis;Coumarin;Anticoagulant;Structure characterization
TQ 201
A
1671-0460(2017)10-1994-04
陕西中医药大学青年基金项目,项目号:2015QN24;咸阳市2016年科学技术研究计划项目,项目号:2016k02-84。
2017-03-11
权彦(1976-),男,陕西省咸阳市人,硕士,2002年毕业于西北大学化学专业,研究方向:药物合成及药效学研究。E-mail:quanyan1@sohu.com。
