高效液相色谱-串联质谱法测定牛奶和鸡肉中4 种激素本底值
2017-11-13孙利东许秀丽聂雪梅许博舟洪云鹤
孙利东,许秀丽,袁 飞,聂雪梅,许博舟,洪云鹤,张 峰,*
(1.中国检验检疫科学研究院食品安全研究所,北京 100176;2.内蒙古满洲里出入境检验检疫局技术中心,内蒙古 满洲里 021400)
高效液相色谱-串联质谱法测定牛奶和鸡肉中4 种激素本底值
孙利东1,2,许秀丽1,袁 飞1,聂雪梅1,许博舟1,洪云鹤1,张 峰1,*
(1.中国检验检疫科学研究院食品安全研究所,北京 100176;2.内蒙古满洲里出入境检验检疫局技术中心,内蒙古 满洲里 021400)
建立牛奶和鸡肉中4 种激素(睾酮、甲基睾酮、孕酮、氢化可的松)本底值同时测定的高效液相色谱-串联质谱方法。样品经β-葡萄糖醛酸酶/芳香基硫酸酯酶水解,加入乙腈超声提取,正己烷除脂肪。采用ZORBAX SB-C18色谱柱分离,以0.1%甲酸溶液和甲醇为流动相梯度洗脱,分别在电喷雾离子源正、负离子模式下以多反应监测模式分段扫描同时检测,基质匹配外标法定量。结果表明,4 种激素在相应的质量浓度范围内线性良好,相关系数均大于0.991;在牛奶中的检出限(RSN=3)和定量限(RSN=10)分别为0.02~0.10 μg/kg和0.06~0.33 μg/kg,在鸡肉中的检出限和定量限分别为0.03~0.12 μg/kg和0.10~0.40 μg/kg;在3 个水平下的平均回收率为牛奶56.0%~132.8%,鸡肉79.6%~122.0%;相对标准偏差为牛奶1.8%~17.3%,鸡肉2.0%~18.1%。该方法简单、快速、准确,适用于对牛奶和鸡肉中4 种激素进行快速筛查和本底值测定。
激素;鸡肉;牛奶;本底值;高效液相色谱-串联质谱法
氢化可的松是人工合成也是天然存在的一类糖皮质激素,主要影响糖、蛋白质和脂肪代谢、提高中枢神经系统兴奋性的作用。睾酮(睾丸酮)是主要的雄性激素,属类固醇激素。甲基睾酮为天然睾酮的17-α位甲基衍生物,具有雄激素和蛋白同化作用。孕酮是一种天然孕激素,又名黄体酮。以上4 种激素具有影响动物性别分化、缩短动物生长周期以及较强的糖代谢及抗炎等作用,可以通过食物链和环境残留对人和动物健康、社会和生态环境造成直接和潜在危害[1]。已有研究表明,过多的激素进入人体可使内分泌失常,导致代谢紊乱或者肿瘤等疾病[2]。欧盟[3]等国际组织及国家均对动物源性食品中激素的残留作出了严格要求。我国也有相关法律规定,禁止一些激素在动物性食品中检出[4]。在现有报道中多是激素多残留同时测定的方法,主要用于牛奶[5-6]、肉类[7-9]、海产品[10]、保健品[11]、化妆品[12]、饲料[13]等方面,还没有专门针对本底值测定的分析方法。并且牛奶和鸡肉中激素本底值水平一直没有相关监控数据,通过技术手段严格监测激素的本底值含量,对于确保食品安全具有极为重要的意义。
本研究针对牛奶和鸡肉中具有代表性的睾酮、孕酮、氢化可的松和甲基睾酮4 种激素本底值含量测定,建立了特异性的样品前处理方法和高效液相色谱-串联质谱分析方法,能够很好地用于测定4 种激素的本底值含量。由于激素分析时基质效应显著,且天然激素含量水平低,对检测方法的灵敏度要求比较高。本研究采用液液萃取净化法对样品中的4 种激素进行提取和净化,选用的溶剂既能达到去除蛋白质等杂质的目的,也无需复杂的仪器和净化装置,使处理过程大大简化,且结果可靠满意,满足痕量分析要求。高效液相色谱串联质谱法因其灵敏度高,选择性和特异性好,能够对低浓度的样品进行良好确认,已成为动物源性食品中痕量兽药残留分析的重要方法。由于氢化可的松和其他3 种性激素在结构上的差异,质谱分析时需要采用不同采集模式,本方法建立了正、负离子源分段采集模式检测4 种激素残留,与现有方法比较提高了灵敏度,缩短分析时间,实现了不同种类激素准确、快速的同时检测,适用于4 种激素在牛奶和鸡肉中本底值的测定。
1 材料与方法
1.1 材料与试剂
牛奶、鸡肉样品 市购;激素标准品:睾酮(纯度99.0%)、17α-甲基睾酮(纯度98.5%)、氢化可的松(纯度99.8%)、孕酮(纯度99.3%) 德国Dr. Ehrenstorfer公司;乙酸、乙酸钠、氯化钠(均为分析纯) 广州化学试剂厂;乙腈、甲醇、正己烷(均为色谱纯) 美国赛尔科技公司;β-葡萄糖醛酸酶/芳香基硫酸酯酶溶液 美国M erck公司;实验用纯水由M illipore纯水仪制备。
1.2 仪器与设备
1260高效液相色谱 美国安捷伦公司;API5500三重四极杆串联质谱仪 美国应用生物系统中国公司;HH-6恒温水浴 江苏金坛亿通电子有限公司;VM-6涡旋振荡器 北京五洲东方科技发展有限公司;KQ-500DE超声仪 上海楚定分析仪器有限公司;Allegra X-22R型高速冷冻离心机 美国Beckman Coulter公司;R-220SE型旋转蒸发仪 瑞士Büchi公司。
1.3 方法
1.3.1 相关溶液的配制
乙酸-乙酸钠缓冲溶液的配制:称取22.9 g五水乙酸钠,加入10 m L乙酸,用水溶解并定容到1 000 m L,用乙酸调节pH 5.2。
标准储备溶液的配制:分别准确称取各激素药物标准品,配制成质量浓度为100 mg/L标准储备液,-18 ℃避光保存。
1.3.2 色谱、质谱条件
色谱条件:A g ilen t ZORBA X SB-C18色谱柱(150 mm×4.6 mm,5 μm);流动相A为0.1%甲酸溶液,流动相B为甲醇;流速0.8 m L/m in;进样量10 μL;梯度洗脱程序:0~2.0 m in,B相30%~85%,2.0~6.5 m in,B相85%~100%,6.5~8.5 m in,B相100%,8.5~9.5 m in,B相100%~30%,9.5~11.5 m in,B相30%。
质谱条件:电喷雾离子源;多反应监测(multip le reaction m onitoring,MRM)采集方式;干燥气温度350 ℃;干燥气流速10.0 L/m in;雾化气压力275.8 kPa(40.0 psi);毛细管电压5 500 V;分辨率:MS1、MS2均为Unit;其他质谱采集参数见表1。

表1 4 种激素质谱分析参数Table 1 M ass spectrometric conditions of 4 hormones
本方法在质谱分析时采用分段扫描,0~6 m in为电喷雾离子源负离子源扫描,6~11.5 m in切换为电喷雾离子源正离子源扫描。
1.3.3 样品前处理
称取样品5.00 g于50 m L离心管中,加入100 μL β-葡萄糖醛酸酶/芳香基硫酸酯酶溶液,10 m L乙酸-乙酸钠缓冲溶液,37 ℃水浴中酶解12 h。取出,降至室温,加入10 m L乙腈,涡旋混匀,超声提取20 m in,水平振荡5 m in,10 000 r/m in离心10 m in,移出乙腈层于另一离心管中。下层水相再加入10 m L乙腈重提1 次,合并2 次乙腈提取液。加入5 m L正己烷,振荡5 m in,4 000 r/m in离心10 m in,移去正己烷层,向乙腈提取液中加入1.0 g NaCl固体,静止分层。将乙腈层移入鸡心瓶中,旋蒸至干。加入1 m L 30%甲醇溶液复溶。过0.22 μm滤膜,待上机测定。
1.3.4 基质匹配标准曲线的绘制
取空白样品经1.3.2节处理,得到空白样品提取液,稀释标准品,得到系列基质匹配标准溶液,上机测定,得到基质匹配标准曲线。
1.3.5 基质效应评定
采用相对响应值法[14]根据下式评定4 种激素的基质效应:

式中:Am为空白基质标准响应值;Ac为纯溶剂标准响应值。
基质效应在质谱检测方法中普遍存在,是影响方法灵敏度的主要因素。当基质效应在80%~120%时,认为基质干扰对检测结果无影响,但当基质效应小于80%或大于120%时,认为基质效应对检测结果的影响不可忽略。本实验分别测定牛奶和鸡肉基质对4 种激素的影响,如表2所示,表明大部分激素的基质效应都在61%~82%之间。所以本研究采用基质匹配标准曲线定量的方法来消除基质效应对检测结果的影响。

表2 4 种激素的基质效应Table 2 M atrix effects of 4 hormones
2 结果与分析
2.1 质谱分析条件优化
在对氢化可的松扫描方式选择时,分别采用电喷雾正离子源和负离子源扫描,对比了相同质量浓度条件下,2 种不同离子源时氢化可的松的响应,负离子源时的响应远大于正离子源时的响应。可能是由于氢化可的松的分子在负离子源条件下[15],易加和甲酸根后形成甲酸去质子的母离子,继而形成失去甲酸的碎片,在仪器分析时响应最强。3 种性激素选用正离子模式扫描,为了达到4 种激素同时检出,在质谱分析时,选用了分段扫描的方式,在0~6 m in时,对氢化可的松进行负模式采集,在6~11.5 m in时,对3 种性激素进行正离子模式采集,如图1所示。
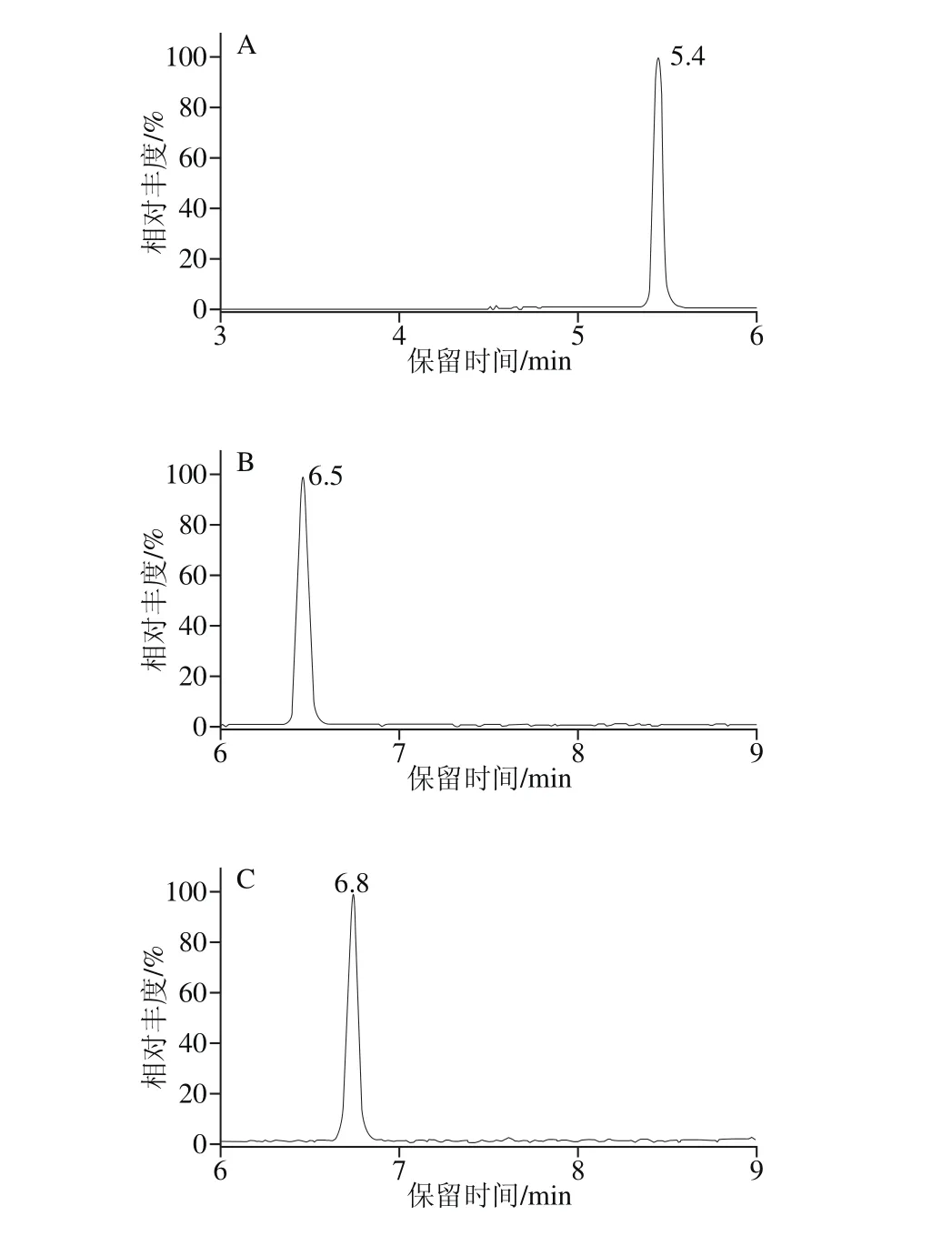
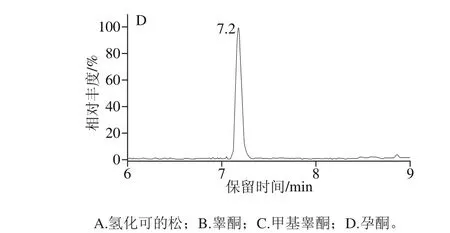
图1 4 种激素的MRM图(5.0 μg/L)Fig. 1 MRM chromatograms of 4 hormones (5.0 μg/L)
2.2 色谱分析条件优化
流动相的选择分别考察了0.1%甲酸溶液+乙腈和0.1%甲酸溶液+甲醇时,4 种激素的分离和响应情况。在这2 种流动相条件下,4 种激素都能达到良好分离。如图2所示,在选用甲醇为有机相时,3 种性激素响应强度远大于选用乙腈时的响应强度,氢化可的松响应受到影响不大,所以选用0.1%甲酸+甲醇为流动相。在水相中加入了0.1%的甲酸后,可大大增强3 种性激素的响应,并且氢化可的松的响应强度并不受很大影响。
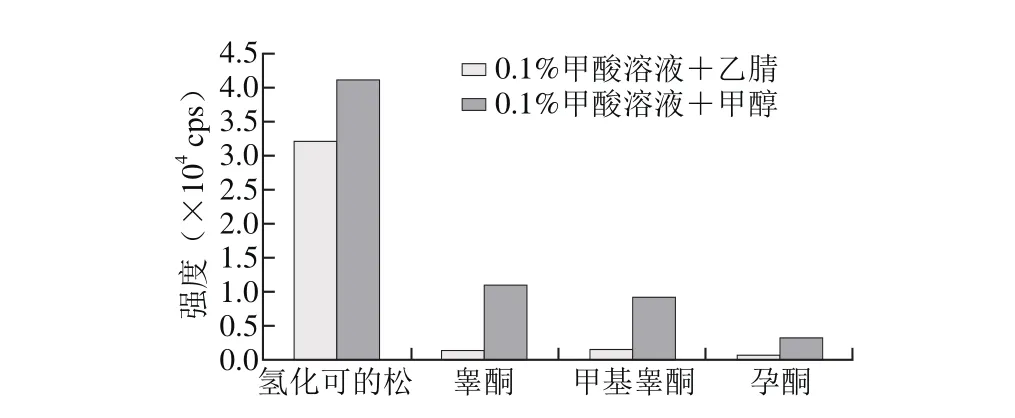
图2 4 种激素在不同流动相时的响应对比Fig. 2 Effect of mobile phase composition on the intensity of response to 4 hormones
2.3 样品前处理条件优化
2.3.1 前处理方法选择

图3 3 种提取净化方法对4 种激素的回收率对比Fig. 3 Effect of cleanup m ethods on the recovery of 4 hormones
如图3所示,分别考察了3 种样品前处理方法,方法1[16]将样品在乙酸铵缓冲溶液中酶解后再经甲醇提取,使用ENV I-Carb和氨基SPE柱净化,由于提取溶液体积接近150 m L,柱净化时间长,大大降低样品前处理效率;方法2[17]是样品在乙酸铵缓冲溶液中酶解,加入甲醇提取后,甲基叔丁基醚萃取,再经过浓缩复溶,SPE柱净化,该方法操作步骤繁琐,增加目标物损失,回收率低。方法3即本实验采用方法,将样品在乙酸铵缓冲溶液中酶解,加入乙腈提取,由于乙腈有很好的除去蛋白作用,可以实现目标化合物与基质有效分离,再用正己烷除脂,即完成了目标的提取与净化,浓缩复溶后即可上机测定。本方法步骤简单高效,净化效果良好,目标物损失较少,在降低基质效应的同时能够保证较高回收率。
2.3.2 提取溶剂选择
分别考察乙腈、乙酸乙酯和甲基叔丁基醚的提取效果[18-25],结果表明乙酸乙酯和甲基叔丁基醚的回收率都不及乙腈,最终选用乙腈用作提取溶剂。由于蛋白质在乙腈中的溶解度很低,在提取过程中,可以很好地除去基质中的蛋白质,再用正己烷除脂净化,便可达到测定要求。牛奶和鸡肉2 种基质的空白样品和加标图谱如图4、5所示。目标物附近没有干扰峰,改进后的样品处理方法能够满足激素本底值测定的要求。
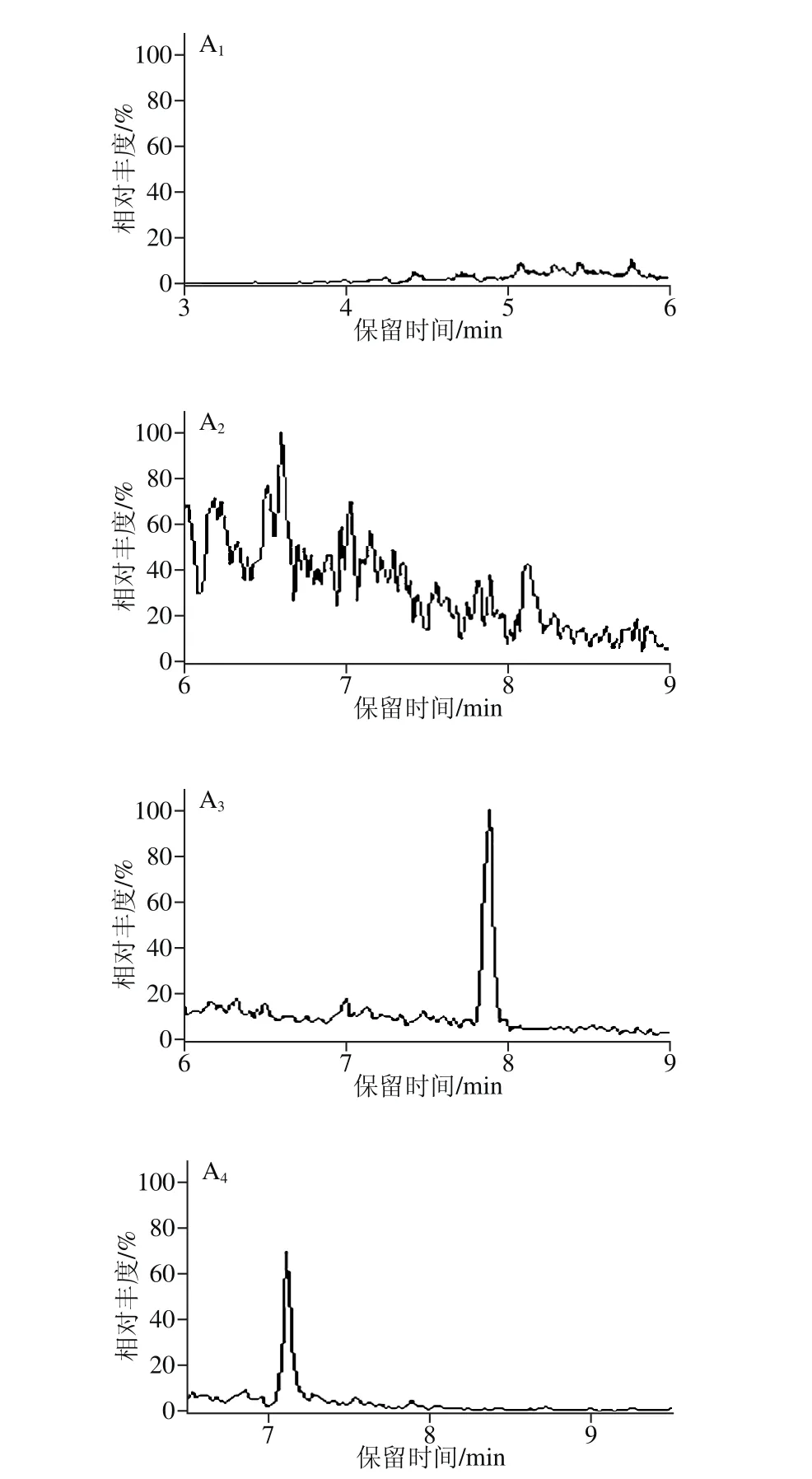
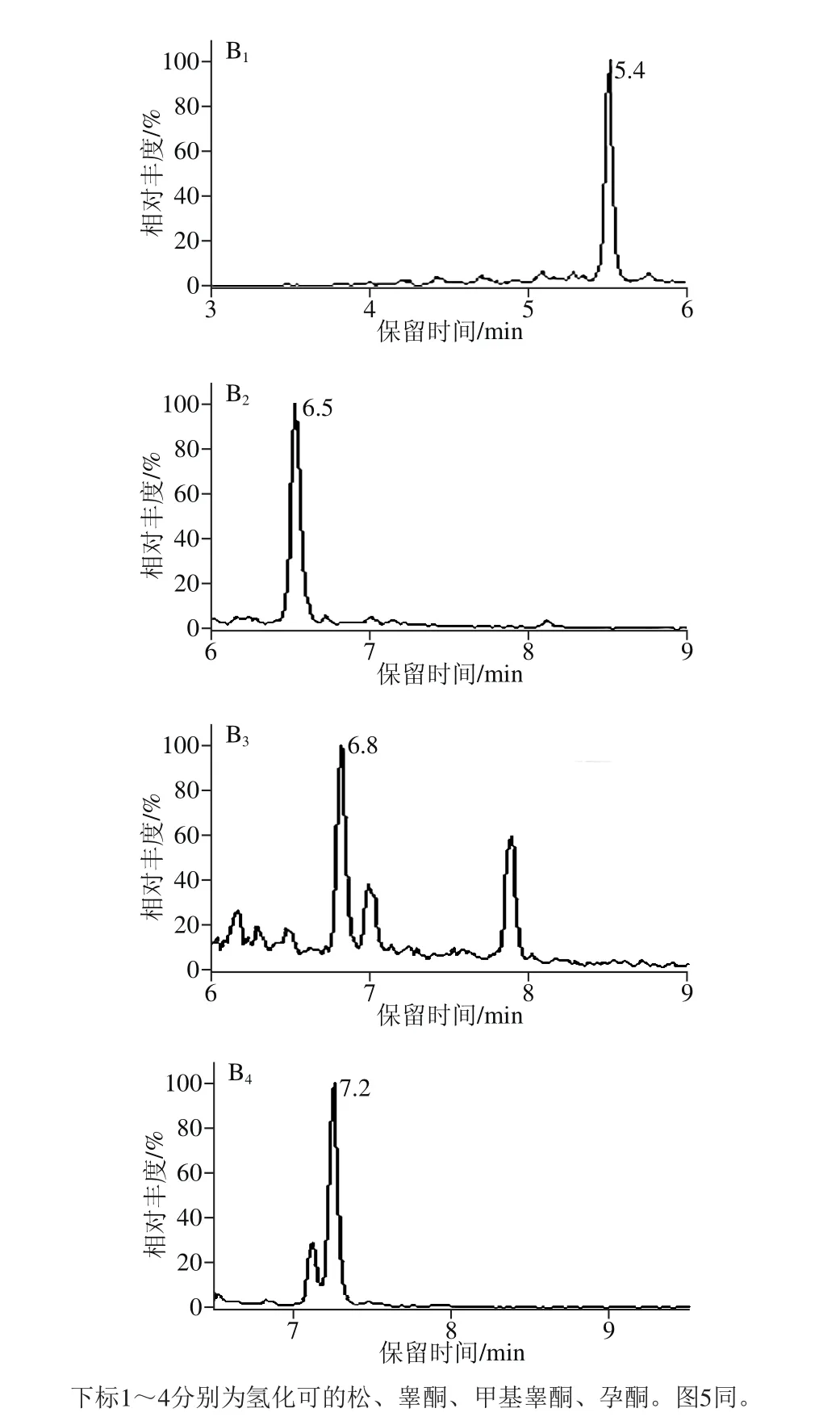
图4 空白牛奶样品(A)和加标牛奶样品(B)的MRM图(加入量1.0 μg/kg)Fig. 4 MRM chromatogram s of blank m ilk sam p le (A) and spiked m ilk sam p le (B) (at 1.0 μg/kg)

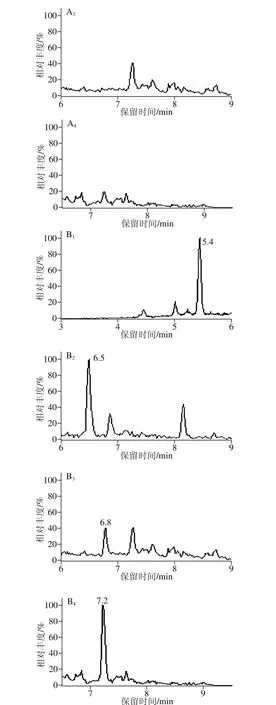
图5 空白鸡肉样品(A)和加标鸡肉样品(B)的MRM图(加入量1.0 μg/kg)Fig. 5 MRM chromatogram s of blank chicken sam p le (A) and spiked chicken sam p le (B) (at 1.0 μg/kg)
2.4 校准曲线、检出限和定量限结果
如表3所示,显示能够满足检测要求。本方法用空白基质提取溶液逐级稀释标准品后测定,根据响应信号的信噪比来确定方法的检出限和定量限[25-31]。当RSN为3时,确定为方法的检出限,当RSN为10时,确定为方法定量限。如表4所示,牛奶和鸡肉的检出限和定量限分别都在0.12 μg/kg和0.40 μg/kg以下,完全可以满足本底值检测要求。

表3 4 种激素的线性方程、相关系数和线性范围Table 3 Linear equations, linear relationships and linear ranges of the 4 hormones
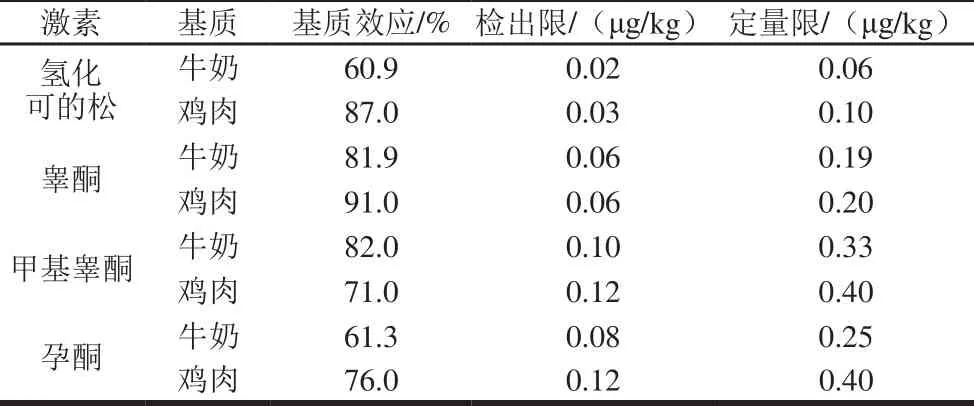
表4 4 种激素的检出限和定量限Table 4 LODs and LOQs of the hormones
2.5 准确度和精密度结果
在样品中分别添加1、2 倍和10 倍定量限的标准溶液,进行测定,如表5所示。牛奶和鸡肉的加标回收率在56.0%~132.8%之间。
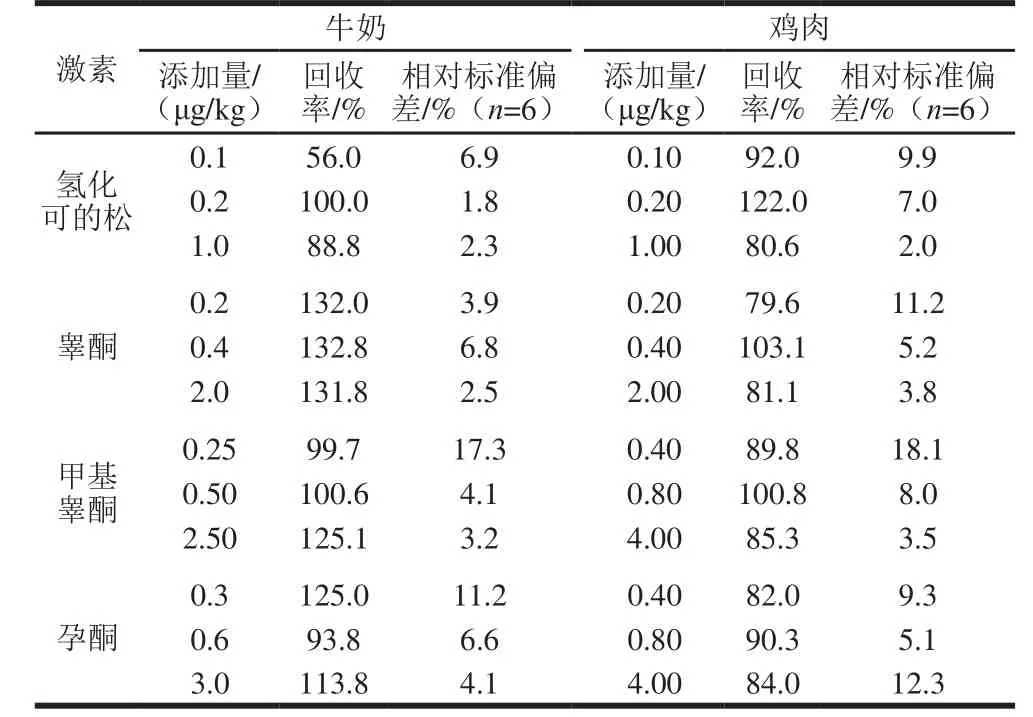
表5 4 种激素在牛奶和鸡肉中的加标回收率和相对标准偏差(n= 6)Table 5 Recoveries and relative standard deviations (n = 6) of the hormones in spiked m ilk and chicken samp les
2.6 实际样品测定
本实验测定了市售11 份牛奶和7 份鸡肉样品中4 种激素的本底值含量。如表6所示,11 份牛奶样品氢化可的松含量在0.14~0.30 μg/kg之间,孕酮含量在0.50~3.34 μg/kg之间;5份鸡肉样品氢化可的松含量在0.06~0.43 μg/kg之间,1 份鸡肉样品中孕酮含量为0.26 μg/kg;牛奶和鸡肉样品中均未检出睾酮和甲基睾酮。实验结果表明,本方法适用于牛奶和鸡肉中4 种激素的本底值检测。
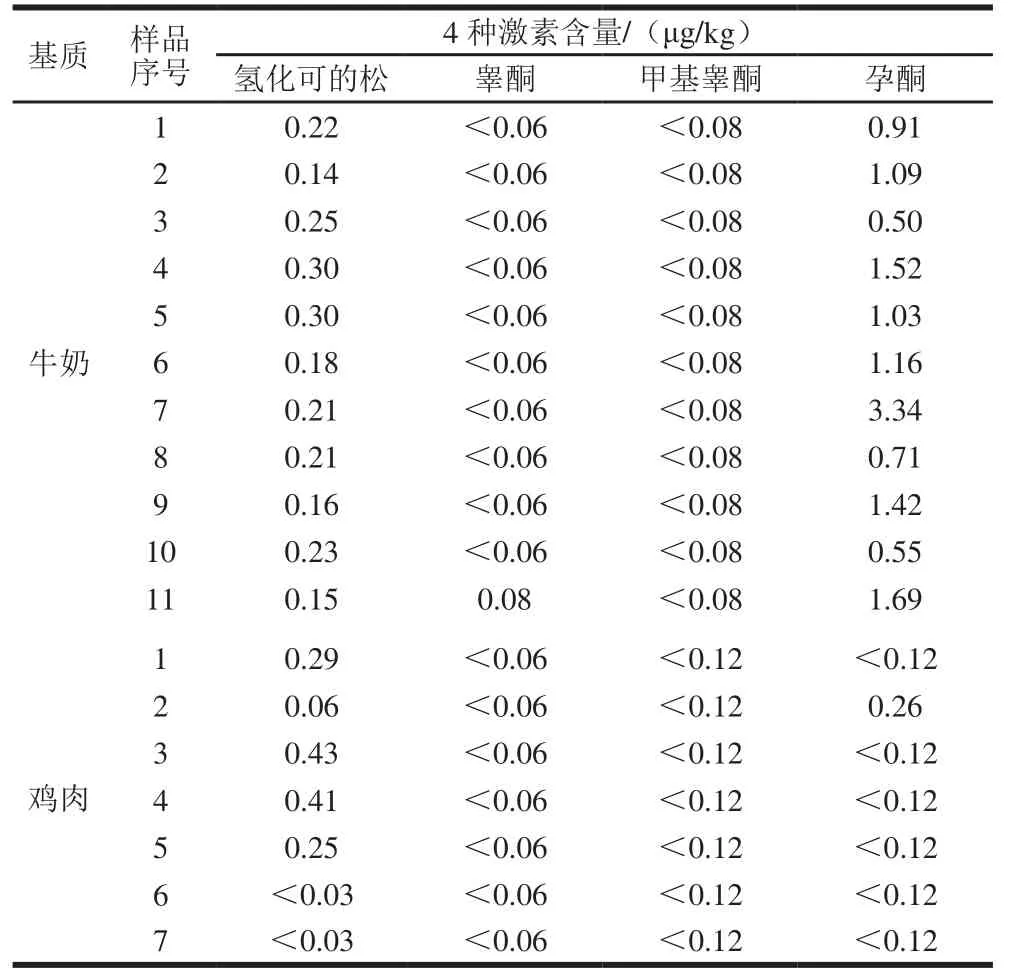
表6 4 种激素在18 份样品中的测定结果Table 6 Contents of 4 hormones in 18 real sam p les
3 结 论
本研究针对牛奶和鸡肉中具有代表性的睾酮、孕酮、氢化可的松和甲基睾酮4 种激素本底值含量测定,通过对样品前处理和仪器分析方法的改进优化,建立了特异性的分析方法。实验结果表明,本方法操作简单、灵敏度高、准确性好、检测成本低,为牛奶和鸡肉中4 种激素的本底值测定提供技术保障。
[1] 李俊锁, 王超. 兽药残留分析[M]. 上海: 上海科学技术出版社, 2002:578-658.
[2] 卫生部疾病预防控制局. 痢疾防治手册[M]. 北京: 人民卫生出版社,2006: 11-12.
[3] European Union (1996). Council Directive 96/22/EC of 29 April 1996 concerning the prohibition on the use in stock farm ing of certain substances having a hormonal or thyrostatic action and of β-agonists, and repealing directives 81/602/EEC, 88/146/ EEC and 88/299/EEC[S].
[4] 农业部. 农业部公告第235号[EB/OL]. (2008-06-29) [2017-01-20].http://www.moa.gov.cn/zw llm/nybz/index_3.htm.
[5] 段永生, 王炳玲, 艾连峰, 等. 在线净化液相色谱-串联质谱法快速测定牛奶中的6 种孕激素[J]. 色谱, 2014, 32(6): 647-652. DOI:10.3724/SP.J.1123.2014.01044.
[6] 李宁, 张玉龙, 林涛, 等. UPLC-MS法同时测定牛奶中磺胺类、喹诺酮类、甾体激素类及四环素类兽药残留[J]. 分析测试学报, 2016,35(6): 714-718. DOI:10.3969/j.issn.1004-4957.2016.06.014.
[7] 牛晋阳, 孙焕, 李莹莹. 高效液相色谱-串联质谱法测定猪肉中10 种类固醇类激素残留[J]. 食品科学, 2010, 31(4): 230-232.
[8] 龚兰, 陈明, 魏娴, 等. 高效液相-电喷雾串联质谱法测定动物肌肉组织中的糖皮质激素[J]. 分析化学, 2016, 44(1): 74-80. DOI:10.11895/j.issn.0253-3820.150561.
[9] 徐锦忠, 张晓燕, 丁涛, 等. 高效液相色谱-串联质谱法同时检测鸡肉和鸡蛋中合成类固醇类激素和糖皮质激素[J]. 分析化学, 2009,37(3): 341-346. DOI:10.3321/j.issn:0253-3820.2009.03.006.
[10] 张爱芝, 王全林, 沈坚, 等. 超高效液相色谱-串联质谱法同时测定鱼制品中残留的7 种性激素[J]. 色谱, 2010, 28(2): 190-196.
[11] 何小琴, 郗存显, 唐柏彬, 等. 超高效液相色谱-串联质谱法快速测定口服液中非法添加的25 种激素[J]. 分析测试学报, 2014, 33(6): 635-641. DOI:10.3969/j.issn.1004-4957.2014.06.003.
[12] 罗辉泰, 黄晓兰, 吴惠勤, 等. 固相萃取/液相色谱-串联质谱法同时测定面膜类化妆品中非法添加的53 种糖皮质激素[J]. 分析测试学报, 2016, 35(2): 119-126. DOI:10.3969/j.issn.1004-4957.2016.02.001.[13] 张峰, 许成保, 蓝芳, 等. 超高效液相色谱-四极杆飞行时间质谱法测定饲料中9 种雄性激素类药物[J]. 分析化学, 2012, 40(1): 101-106.DOI:10.3724/SP.J.1096.2012.10419.
[14] 刘宏程, 李宁, 林涛, 等. 基质固相分散-超高效液相色谱-质谱检测器测定牛奶中9 种类固醇激素残留[J]. 色谱, 2015, 33(11): 1163-1168. DOI:10.3724/SP.J.1123.2015.06035.
[15] 张协光, 郑彦婕, 曾泳艇, 等. 液相色谱-质谱联用测定食品中19 种激素[J]. 食品工业, 2016(2): 281-286.
[16] 国家质量监督检验检疫总局. 动物源性食品中激素多残留检测方法液相色谱-质谱/质谱法: GB/T 21981—2008[S]. 北京: 中国标准出版社, 2008.
[17] 国家质量监督检验检疫总局. 牛肝和牛肉中睾酮、表睾酮、孕酮残留量测定液相色谱-串联质谱法: GB/T 20758—2006[S]. 北京:中国标准出版社, 2006.
[18] GOYON A, CAI J Z, KRAEHENBUEHL K, et al. Determ ination of steroid hormones in bovine m ilk by LC-MS/MS and their levels in Sw iss Holsterin cow milk[J]. Food Additives & Contam inants: Part A,2016, 33(5): 804-816. DOI:10.1080/19440049.2016.1175186.
[19] WANG G, HSIEH Y, CUI X, et al. U ltra-performance liquid chrom atography/tandem mass spectrom etric determ ination o f testosterone and its metabo lites in in vitro sam p les[J]. Rapid Communications in Mass Spectrometry, 2006, 20(14): 2215-2221.DOI:10.1002/rcm.2580.
[20] GUO T, TAYLOR R L, SINGH R J, et al. Simultaneous determ ination of 12 steroids by isotope dilution liquid chromatography-photospray ionization tandem mass spectrometry[J]. Clinica Chim ica Acta, 2006,372(1): 76-82. DOI:10.1016/j.cca.2006.03.034.
[21] VANDERFORD B J, PEARSON R A, REXING D J, et al. Analysis of endocrine disruptors, pharmaceuticals, and personal care products in water using liquid chromatography/tandem mass spectrometry[J]. Analytical Chem istry, 2003, 75(22): 6265-6274. DOI:10.1021/ac034210g.
[22] VANDERFORD B J, SNYDER S A. Analysis of pharmaceuticals in water by isotope dilution liquid chromatography/tandem mass spectrometry[J]. Environmental Science & Technology, 2006, 40(23):7312-7320.
[23] WU Z, ZHANG C, YANG C, et al. Simu ltaneous quantitative determ ination of norgestrel and progesterone in human serum by highperformance liquid chromatography-tandem mass spectrometry w ith atmospheric pressure chem ical ionization[J]. Analyst, 2000, 125(12):2201-2205.
[24] TÖLGYESI Á, VEREBEY Z, SHARMA V K, et al. Simultaneous determ ination of corticosteroids, androgens, and p rogesterone in river w ater by liqu id chrom atography-tandem m ass spectrometry[J]. Chemosphere, 2010, 78(8): 972-979. DOI:10.1016/j.chemosphere.2009.12.025.
[25] GUO T, CHAN M, SOLDIN S J. Steroid profiles using liquid chromatography-tandem mass spectrometry w ith atmospheric pressure photoionization source[J]. A rchives o f Patho logy &Laboratory M edicine, 2004, 128(4): 469-475. DOI:10.1043/1543-2165(2004)128<469:SPULCM>2.0.CO;2.
[26] TA I S S C, BEDNER M, PH INNEY K W. Development of a candidate reference measurement procedure for the determ ination of 25-hydroxyvitam in D3 and 25-hydroxyvitam in D2 in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry[J]. Analytical Chem istry, 2010, 82(5): 1942-1948.DOI:10.1021/ac9026862.
[27] KUSHNIR M M, ROCKWOOD A L, ROBERTS W L, et al. Liquid chromatography tandem mass spectrometry for analysis of steroids in clinical laboratories[J]. Clinical Biochem istry, 2011, 44(1): 77-88.DOI:10.1016/j.clinbiochem.2010.07.008.
[28] GOSETTI F, MAZZUCCO E, GENNARO M C, et al. Ultra high performance liquid chromatography tandem mass spectrometry determ ination and profi ling of prohibited steroids in human biological matrices. A review[J]. Journal of Chromatography B, 2013, 927: 22-36. DOI:10.1016/j.jchromb.2012.12.003.
[29] 赵超敏, 岳振峰, 吴晖, 等. 黄油中8 种类固醇激素的液相色谱/串联质谱检测[J]. 分析化学, 2014, 42(3): 360-366. DOI:10.3724/SP.J.1096.2014.30857.
[30] 祝伟霞, 刘亚风, 袁萍, 等. 液相色谱-串联质谱法快速测定婴幼儿配方奶粉中39 种激素残留量[J]. 色谱, 2010, 28(11): 1031-1037.DOI:10.3724/SP.J.1123.2010.01031.
[31] 邹红梅, 左舜宇, 黄东仁, 等. 高效液相色谱-串联质谱法同时测定水产品中雄激素和糖皮质激素残留[J]. 中国渔业质量与标准, 2016,6(2): 45-50.
High Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of Background Values of 4 Hormones in M ilk and Chicken
SUN Lidong1,2, XU Xiuli1, YUAN Fei1, NIE Xuemei1, XU Bozhou1, HONG Yunhe1, ZHANG Feng1,*
(1. Institute of Food Safety, Chinese Academy of Inspection and Quarantine, Beijing 100176, China;2. Technology Center of the Manzhouli Entry-Exit Inspection and Quarantine Bureau, Manzhouli 021400, China)
A high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method was developed for the simultaneous determ ination of background values of 4 hormones (testosterone, methyltestosterone, progesterone and hydrocortisone) in m ilk and chicken. The samples were hydrolyzed by β-glucuronidase and ultrasonically extracted w ith acetonitrile, and then the extract was defatted w ith hexane. The analytes were seperated on a ZORBAX SB-C18column w ith methanol-0.1% form ic acid as mobile phase by gradient elution. The mass spectrometer was run in multiple reaction monitoring mode (MRM) using positive- and negative-ion electrospray ionization (ESI), respectively. The quantitative analysis was carried out by matrix-matched external standard method. The results showed that the calibration curves had good linearity for 4 hormones in the tested concentration ranges, w ith correlation coefficients of more than 0.991. The lim its of detection (LOD, RSN= 3) and lim its of quantitation (LOQ, RSN= 10) were 0.02–0.10 and 0.06–0.33 μg/kg in m ilk, and 0.03–0.12 μg/kg and 0.10–0.40 μg/kg in chicken, respectively. The average recoveries at three spiked levels 56.0%–132.8% for m ilk and 79.6%–122.0% for chicken, and the precisions expressed as relative standard deviations (RSDs)were 1.8%–17.3% for milk and 2.0%–18.1% for chicken. The method proved to be simple, rapid, accurate, and suitable for rapid screening and detection of background values for 4 hormones in m ilk and chicken.
hormones; chicken; m ilk; background values; high performance liquid chromatography-tandem mass spectrometry
10.7506/spkx1002-6630-201722043
2017-01-24
“十三五”国家重点研发计划重点专项(2016YFD0401103;2016YFF0203903)
孙利东(1983—),女,工程师,硕士,研究方向为食品中有毒有害物质、兽药残留检测。
E-mail:sunlidong1983@sina.com
*通信作者:张峰(1974—),男,研究员,博士,研究方向为分析化学、食品安全。E-mail:fengzhang@126.com
O658
A
1002-6630(2017)22-0291-07
孙利东, 许秀丽, 袁飞, 等. 高效液相色谱-串联质谱法测定牛奶和鸡肉中4 种激素本底值[J]. 食品科学, 2017, 38(22):291-297. DOI:10.7506/spkx1002-6630-201722043. http://www.spkx.net.cn
SUN Lidong, XU Xiuli, YU AN Fei, et al. High performance liquid chromatography-tandem mass spectrometry method for simultaneous determ ination of background values of 4 hormones in m ilk and chicken[J]. Food Science, 2017, 38(22):291-297. (in Chinese w ith English abstract)
10.7506/spkx1002-6630-201722043. http://www.spkx.net.cn
