茂基稀土邻氨基苯甲酰胺基双负离子配合物的合成及其环胺羰化反应和脒基化反应
2017-11-13刘瑞婷翁林红周锡庚
孙 燕 刘瑞婷 翁林红 周锡庚*,,2
茂基稀土邻氨基苯甲酰胺基双负离子配合物的合成及其环胺羰化反应和脒基化反应
孙 燕1刘瑞婷1翁林红1周锡庚*,1,2
(1复旦大学化学系,上海市分子催化与功能材料重点实验室,上海 200433)
(2金属有机化学国家重点实验室,上海 200032)
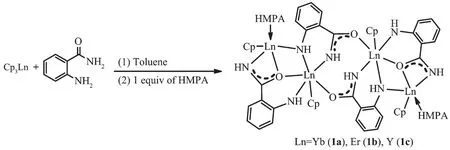
Cp3Ln与邻氨基苯甲酰胺在甲苯中反应,之后在HMPA和甲苯中结晶,以中等到高收率得到四核稀土有机配合物[CpLn(μη2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)LnCp(HMPA)}2(Ln=Yb,1a;Er,1b;Y,1c)。 化合物 1 与 4 倍物质的量的 PhNCO 在甲苯中反应形成 1,3-喹唑啉二氧基(Quo)双负离子稀土配合物[Cp2Ln(μ3-η2∶η2∶η1-Quo)]3Ln(HMPA)2(Ln=Yb,2a;Er,2b;Y,2c),表明化合物1中的Ln-NHAr键和ArCONH-Ln键能与异氰酸酯分子发生连续加成/胺消除反应,形成1,3-喹唑啉二氧基骨架。但化合物1a~1c 与iPrN=C=NiPr反应,仅得到 ArNH 基单加成产物{Cp2Ln[μ-η1∶η1∶η2-iPrNC(NHiPr)NC6H4CONH]}3Ln(HMPA)3(Ln=Yb,3a;Er,3b;Y,3c)。 而 Cp3Ln 与邻氨基苯甲酰胺和iPrN=C=NiPr在甲苯中进行“一锅”反应,则形成双核配合物{CpLn[μ-η1∶η2∶η2-NHCOC6H4NC(NHiPr)NiPr]}2(Ln=Yb,4a;Er,4b;Y,4c)。 值得注意的是,HMPA 能够诱导配合物 4 发生配体重排反应,转化成化合物 3。
稀土配合物;环胺羰化反应;异氰酸酯;邻氨基苯甲酰胺;加成反应
0 Introduction
There is currently a fundamental interest in the reactivity of organolanthanide complexes toward unsaturated small molecules,because this is the source for design of new catalytic reactions and catalysts[1-6].The selective insertion of an organic functional group into a lanthanide-ligand bond has proved to be a successful and pervasive method for the formation of carbon-carbon and carbon-heteroatom bonds in synthesis of organolanthanide derivatives and organic compounds[6-13].For example,by applying this type of insertions as the key step,many organolanthanidecatalyzed reactions such as hydrosilylation[7],hydroamination[2b,8], hydroalkoxylation[9], hydrothiolation[10],hydrophosphination[11],hydroboration[12],and C-H bond addition[2c,13]of diverse C-C unsaturated substrates have been developed.In contrast,the development of metathesisreactionsoftrivalentorganolanthanide complexes with unsaturated substrates leading to the complete cleavage of the unsaturated chemical bond,lags far behind,despite their fundamental scientific interest and the potential utility in organic synthesis.The major reason might be attributed to the absence of conventional oxidative-addition/reductive-elimination processes and the difficulties encountered in the formation of rare earth metal-carbon and rare earth metal-heteroatom multiply-bonded intermediates,which often involvein transition metal-catalyzed metathesis reactions of unsaturated substrates[14].Recent pioneering works show that reaction of tetranuclear rare earth metal polyhydrido complexes with CO yielded ethylene and the corresponding tetraoxo cubane complexes[15a].Chen and coworkers reported that yttrium dihydride reacted with Ph3P=Se to give an yttrium selenide complex in the company of the liberation of Ph3P and H2[15b].Furthermore,methylidene complexes of rare earth metals have proven their ability to cleave the C=O,N=N and C=N double bonds[16].Despite these achievements,the use of trivalent organolanthanide complexes as reagents for the transformation of an unsaturated functional group into another unsaturated one in the context of metathesis reactionsremainsunderutilized.The question of whether or not rare earth metal amido complexes can promote formation and transformation of unsaturated carbon-heteroatom bonds as readily as rare earth metal polyhydrido and methylidene complexes mediate C=O bond metathesis reaction is still an open issue.
The chemical nature of isocyanates,with an electrophilic center at carbon and nucleophilic centers at oxygen and nitrogen,allows a diverse variety of reactions to occur,thus leading to a great potential for constructing higher organic structures[17].The selectivity ofisocyanatetransformationshasbeen profitably controlled by metal species,since their participation allows the fine-tuning of bond forming processes and reactivity of isocyanates[18].In attempts to establish more selective catalystsystemsand tohave abetter understanding of the mechanistic aspects as well as to develop new reactions,extensivestudieson the reaction of organometallic complexes with isocyanates have been carried out.Noticeably,in contrast to transition metals,selective C=N or C=O bond cleavage of isocyanates mediated by rare earth metals remains a significant challenge,mainly due to the instability of the resulting nitrene and carbonyl complexes and to the facile C=O addition pathways available[18].The stoichiometric reaction of trivalent organolanthanide compounds with isocyanates gives generally the addition products (Scheme 1)[19].

Scheme 1
On the other hand,using isocyanates as CO precursors in organic synthesis remains little explored[20].Hou and coworkers reported in 2004 that yttrocene tetrahydrido complex [(C5Me4SiMe3)Y(μ-H)]4(L)(L=Me3SiCCHCHCSiMe3)could abstract the oxygen from aryl isocyanate to form the corresponding μ3-oxo complex [(C5Me4SiMe3)Y]4(μ3-O)(μ-H)2(L)or [(C5Me4SiMe3)Y]4(μ3-O)2(L),depending on the substrate ratio[21].In the course of our ongoing studies involving the reaction of organolanthanide complexes with isocyanates,we found that the further interaction of the neighboring NH2group with an isocyanate unit inserted into the lanthanide-sulfur bond could lead to unique reactivity and selectivity trends,allowing a mild and efficient construction of the coordinated benzothiazole 2-oxide ligand[20a](Scheme 2).So far,no general method for rare earth metal-based carbonylation has been reported.Given that the above reaction provides an alternative route to carbonylation of organic substrates,we were interested in broadening the reaction.Herein,we report the synthesis and structure of lanthanocene complexes containing oaminobenzamido dianion ligand,and a new reactivity pattern ofamido complexes toward isocyanates:carbonylative coupling/cyclization,which provides a new method for the construction of a coordinated quinazolyldiolate ring skeleton.

Scheme 2
1 Experimental
1.1 General remarks
Alloperations involving air-and moisturesensitive compounds were carried out under an inert atmosphere of purified nitrogen using standard Schlenk techniques.All organic solvents such as THF,toluene,and n-hexanewererefluxed and distilled over sodium benzophenone ketyl under N2prior to use.o-Aminobenzamide,HMPA (hexamethylphosphorictriamide),N,N′-diisopropyl carbodiimide(DIC)and phenyl isocyanate (PhNCO)were purchased from commercial sources and were used without further purification.Elemental analyses for C,H,and N were carried out by using a Rapid CHN-O analyzer.Metal analyses were accomplished using the literature method[22].Infrared spectra for air-and moisturesensitive compounds were obtained on a Nicolet FTIR 360 spectrometer with samples prepared as Nujol mulls.1H NMR data were obtained on a Bruker DMX-400 NMR spectrometer.
1.2 Preparation
1.2.1 Synthesis of[CpYb(μ-η2∶η2-NHC6H4CONH)(μ3-
η1∶η1∶η2-NHC6H4CONH)YbCp(HMPA)]2(1a)To a toluene solution (20 mL)of Cp3Yb (0.331 g,0.898 mmol)was added o-aminobenzamide (0.122 g,0.898 mmol)atroom temperature.Thereaction mixture was stirred at room temperature overnight.Then,HMPA (0.161 g,0.898 mmol)was added to the resulting yellow turbid solution.After stirring for 2 h,the reaction mixture was changed to a clear solution.The solution was then cooled at-10℃overnight to give 1a as orange crystals.Yield:0.388 g (94% ).Anal.Calcd.for C60H80N14O6P2Yb4(%):C,39.01;H,4.36;N,10.61;Yb,37.47.Found(%):C,39.12;H,4.41;N,10.53;Yb,37.33.IR (Nujol,cm-1):3 365 m,3 309 w,1 601 m,1 526 m,1 457 vs,1 378 m,1 327 m,1 145 s,990 s,873 w,758 s.
1.2.2 Synthesis of[CpEr(μ-η2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)ErCp(HMPA)]2(1b)
Following the described method for synthesis of 1a,using Cp3Er (0.496 g,1.37 mmol),o-aminobenzamide (0.187 g,1.37 mmol)and HMPA (0.246 g,1.37 mmol)afforded 1b as light pink crystals.Yield:0.534 g (86%).Anal.Calcd.for C60H80N14O6P2Er4(%):C,39.50;H,4.42;N,10.75;Er,36.67.Found(%):C,39.55;H,4.43;N,10.69;Er,36.58.IR (Nujol,cm-1):3 365 m,3 310 w,1 600 m,1 525 m,1 457 vs,1 379 m,1 327 m,1 144 s,989 s,872 w,757 s.
1.2.3 Synthesis of[CpY(μ-η2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)YCp(HMPA)]2(1c)
Following the described method for synthesis of 1a,using Cp3Y (0.86 g,1.01 mmol),o-aminobenzamide (0.137 g,1.01 mmol)and HMPA (0.181 g,1.01 mmol)afforded 1c as a light yellow crystalline powder.Yield:0.281 g (74%).Anal.Calcd.for C60H80N14O6P2Y4(%):C,47.69;H,5.34;N,12.98;Y,23.54.Found(%):C,47.74;H,5.37;N,12.90;Y,23.52.IR (Nujol,cm-1):3 364 m,3 309 w,1 600 m,1 524 m,1 458 vs,1 378 m,1 327 m,1 143 s,988 s,758 s.1H NMR (C6D6,400 MHz,25 ℃):δ 7.92 (s,NH,2H),7.90 (s,NH,2H),7.08~6.43 (m,C6H4,16H),6.29 (s,C5H5,20H),3.71 (s,NH,2H),3.56 (s,NH,2H),2.28 (d,J=8.0Hz,N(CH3)2,36H).
1.2.4 Synthesis of[Cp2Yb(μ3-η2∶η2∶η1-Quo)]3Yb(HMPA)2(2a)
To a toluene solution (20 mL)of 1a (0.412 g,0.225 mmol)was added PhNCO (0.107 g,0.901 mmol)at 0 ℃.The reaction mixture was stirred at room temperature overnight. The solution was concentrated and cooled at-15℃for several days to give 2a as a yellow powder.Crystals of 2a suitable for X-ray analysis were obtained by recrystallization in a mixed solvent of toluene and THF.Yield:0.230 g(53%).Anal.Calcd.for C66H78N12O8P2Yb4(% ):C,41.25;H,4.09;N,8.75;Yb,36.02.Found (%):C,41.29;H,4.14;N,8.69;Yb,35.96.IR(Nujol,cm-1):3 411 w, 3 339 s,2 192 m,1 598 m,1 537 s,1 465 s,1 373 s,1 300 m,1 149 m,991 s,751 s.
1.2.5 Synthesis of[Cp2Er(μ3-η2∶η2∶η1-Quo)]3Er(HMPA)2(2b)
Following the method described for synthesis of 2a,using 1b (0.552 g,0.30 mmol)and PhNCO (0.144 g,1.21 mmol)afforded 2b as a pink powder.Yield:0.268 g (46%).Anal.Calcd.For C66H78N12O8P2Er4(%):C,41.76;H,4.14;N,8.85;Er,35.24.Found(%):C,41.47;H,4.04;N,8.69;Er,35.16.IR (Nujol,cm-1):3 410 w,3 339 s,2 191 m,1 597 m,1 537s,1 463 s,1 373 s,1 300 m,1 149 m,991 s,753 s.
1.2.6 Synthesis of[Cp2Y(μ3-η2∶η2∶η1-Quo)]3Y(HMPA)2(2c)
Following the method described for synthesis of 2a,using 1c (0.408 g,0.27 mmol)and PhNCO (0.128 g,1.08 mmol)afforded 2c as a light yellow powder.Yield:0.214 g (50%).Anal.Calcd.for C66H78N12O8P2Y4(%):C,50.01;H,4.96;N,10.60;Y,22.44.Found:C,49.96;H,4.91;N,10.49;Y,22.39.IR (Nujol,cm-1):3 407 w,3 337 s,2 192 m,1 597 m,1 536s,1 463 s,1 373 s,1 301 m,1 240 w,1 147 m,990 s,756 s.1H NMR (C6D6,400 MHz,25 ℃): δ 7.34~7.08 (m,C6H4,12H),6.15 (s,C5H5,30H),2.12 (s,NMe2,36H).
1.2.7 Synthesis of{Cp2Yb[μ-η1∶η1∶η3-iPrNC(NHiPr)NC6H4-CONH]}3Yb(HMPA)3(3a)
To a toluene solution (20 mL)of 1a (0.412 g,0.225 mmol)was added DIC (0.141 g,1.12 mmol)at 0℃.The reaction mixture was stirred at room tempera-ture overnight.The solution was concentrated and cooled at-15℃for several days to give 3a as a yellow powder.The yellow crystals of 3a·THF suitable for X-ray analysis were obtained by recrystallization in a mixed solvent of toluene and THF.Yield:0.389 g(70%).Anal.Calcd.for C90H144N21O6P3Yb4(%):C,45.01;H,6.04;N,12.25;Yb,28.82.Found(%):C,45.14;H,6.07;N,12.17;Yb,28.75.IR (Nujol,cm-1):3 410 m,3 381 w,3 280 w,1 600 w,1 522 w,1 460 vs,1 377 s,1 127 m,837 w,727 s.
1.2.8 Synthesis of{Cp2Er[μ-η1∶η1∶η3-iPrNC(NHiPr)NC6H4-CONH]}3Er(HMPA)3(3b)
Following the described method for synthesis of 3a,using 1b (0.552 g,0.30 mmol)and DIC (0.152 g,1.20 mmol)afforded 3b·THF as light pink crystals.Yield:0.375 g (50%).Anal.Calcd.for C90H144N21O6P3Er4(%):C,45.45;H,6.10;N,12.37;Er,28.13.Found(%):C,45.44;H,6.13;N,12.31;Er,28.05.IR(Nujol,cm-1):3 409 m,3 381 w,3 279 w,1 599 w,1 556 w,1 459 vs,1 379 s,1 158 w,1 126 m,988 w,729 s.
1.2.9 Synthesis of{Cp2Y[μ-η1∶η1∶η3-iPrNC(NHiPr)NC6H4-CONH]}3Y(HMPA)3(3c)
Following the described method for synthesis of 3a,using 1c (0.468 g,0.31 mmol)and DIC (0.154 g,1.22 mmol)afforded 3c as light yellow crystalline solid.Yield:0.285 g (43%).Anal.Calcd.for C90H144N21O6P3Y4(%):C,52.35;H,7.03;N,14.25;Y,17.22.Found(%):C,52.39;H,7.09;N,14.23;Er,17.20.IR(Nujol,cm-1):3 406 m,3 380 w,3 280 w,1 598 w,1 554 w,1 460 vs,1 377 s,1 155 w,1 128 m,993 w,726 w.1H NMR(C6D6,400 MHz,25 ℃):δ 8.31 (s,NH,3H),7.23~6.99(m,C6H4,12H),6.09 (s,C5H5,30H),3.75 (m,CH(CH3)2,3H),3.55 (s,NH,3H),3.33 (m,CH(CH3)2,3H),2.49(d,J=8.0 Hz,N(CH3)2,54H),1.34 (d,J=6.4 Hz,CH(CH3)2,18H),0.93 (d,J=6.4 Hz,CH(CH3)2,18H).Crystals of 3c·THF suitable for X-ray analysis were obtained by layering n-hexane to its concentrated toluene/THF solution.
1.2.10 Synthesis of{CpYb[μ-η1∶η2∶η2-iPrNC(NHiPr)NC6H4-CONH]}2(4a)
To a toluene solution (20 mL)of Cp3Yb (0.238 g,0.646 mmol)was added o-aminobenzamide (0.088 g,0.646 mmol)at room temperature.After stirred at room temperature overnight,DIC (0.082 g,0.646 mmol)was added to the resulting yellow turbid solution at 0℃.Then,the reaction mixture was heated at 110 ℃for 12 h.The hot clear solution was slowly cooled to room temperature,affording 4a as yellow crystals.Yield:0.300 g (93%).Anal.Calcd.for C38H50N8O2Yb2(%):C,45.78;H,5.06;N,11.24;Yb,34.71.Found(%):C,45.84;H,5.02;N,11.20;Yb,34.65.IR(Nujol,cm-1):3 396 m,3 381 w,1 598 w,1 463 vs,1 377 s,1 327 m,1 170 m,1 124 w,875 w,763 s.
1.2.11 Synthesis of{CpEr[μ-η1∶η2∶η2-iPrNC(NHiPr)NC6H4-CONH]}2(4b)
Following the method described for synthesis of 4a,using Cp3Er (0.326 g,0.898 mmol),o-aminobenzamide (0.123 g,0.898 mmol)and DIC (0.113 g,0.898 mmol)afforded 4b as light pink crystals.Yield:0.400 g (91%).Anal.Calcd.for C38H50N8O2Er2(%):C,46.32;H,5.11;N,11.37;Er,33.95.Found(%)C,46.39;H,5.15;N,11.28;Er,33.82.IR (Nujol,cm-1):3 399 m,3 379 w,1 598 w,1 548 w,1 459 vs,1 377 s,1 172 m,1 124 m,874 w,764 s.
1.2.12 Synthesis of{CpY[μ-η1∶η2∶η2-iPrNC(NHiPr)NC6H4CO-NH]}2(4c)
Following the method described for synthesis of 4a,using Cp3Y (0.317 g,1.11 mmol),o-aminobenzamide(0.152 g,1.11 mmol)and DIC (0.140 g,1.11 mmol)afforded 4c as light yellow crystals.Yield:0.321 g(70%).Anal.Calcd.for C38H50N8O2Y2(%):C,55.08;H,6.08;N,13.52;Y,21.46.Found(%):C,55.09;H,6.12;N,13.50;Y,21.45.IR (Nujol,cm-1):3 399 m,3 375 w,1 741 w,1 598 w,1 547 w,1 460 vs,1 377 s,1 171 m,1 124 m,763 s.1H NMR (C6D6,400 MHz,25 ℃):δ 7.28~7.11 (m,C6H4,8H),6.94 (s,NH,2H),6.45 (s,C5H5,10H),6.12 (s,NH,2H),3.35~3.22(m,CH(CH3)2,4H),1.28 (d,J=6.4 Hz,CH3,6H),0.86(d,J=6.4 Hz,CH3,6H).
1.2.13 Transformation from 4a to 3a
To a toluene solution (20 mL)of 4a (0.254 g,0.255 mmol)was added HMPA (0.091 g,0.510 mmol)at room temperature.After stirring for 12 h,the solution was concentrated and cooled at-15℃for several days to give 3a as yellow crystals.Yield:0.165 g (54%).
1.3 X-ray data collection,structure determination and refinement
Suitable single crystals were sealed under N2in thin-walled glass capillaries forX-ray structural analysis.X-ray diffraction data were collected on a SMART APEX CCD diffractometer(graphite-monochromated Mo Kα radiation,φ-ω scan technique,λ=0.071 073 nm).The intensity data were integrated by means of the SAINT program[23].SADABS[24]was used to perform area-detector scaling and absorption corrections.The structures were solved by direct methods and were refined against F2using all reflections with the aid of the SHELXTL package[25].All non-hydrogen atoms were found from the difference Fourier syntheses and refined anisotropically.The H atoms were included in calculated positions with isotropic thermal parameters related to those of the supporting carbon atoms but were not included in the refinement.All calculations were performed using the Bruker Smart program.Details of this SQUEEZE are given in the cif files.Crystal data,data collection,and processing parameters for the complexes are summarized in Table 1~2.
CCDC:951003,1a;951104,1b;951102,2a;951004,3a;951106,3b;951005,3c;951103,4a;951107,4b;951105,4c.

Table 1 Crystal and data collection parameters of complexes 1a,1b and 4a~4c

Table 2 Crystal and data collection parameters of complexes 2a and 3·THF
2 Results and discussion
2.1 Syntheses and characterization of lanthanocene derivatives with the o-aminobenzamido dianion ligand
Considering thatthe ureido resulting from addition of amine to isocyanate might potentially undergo the nucleophilic substitution or addition[26-27](Scheme 3),our interest in the rare earth metalmediated C=N bond cleavage of isocyanates prompted us to explore the possibility of reacting two coordinated NH anions with one isocyanate molecule.

Scheme 3
On the basis of this idea,we focused our attention on using the linked NH/CONH dianion lanthanide complexes as the template of the target reaction as the anionic NH and CONH groups offer a degree of selectivity,in which the two NH groups can react either with a reagent having two functionalities or with two separate reagents bearing the same or different functionality.Thus,it may be expected that the different reactivity between coordinated NH and CONH anions toward the same functionality would control reaction degree of advancement,for less reactive substrates only the NH addition takes place,whereas the more reactive ones lead to the occurrence of a tandem reaction involved the two kinds of amido groups.Three o-aminobenzamido dianion lanthanide complexes [CpLn(μ-η2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)LnCp(HMPA)]2(Ln=Yb,1a;Er,1b;Y,1c)as the designed starting materials were synthesized in good yields by protonolysis of Cp3Ln with oaminobenzamide followed by crystallization in a HMPA and toluene mixture (Scheme 4).
Complexes 1a~1c are air-and moisture-sensitive,and readily dissolved in toluene and THF.They are thermally stable at room temperature.Complexes 1a~1c were characterized by elemental analysis and spectroscopic methods.In the IR spectra,all of them show two sharp N-H stretching vibration peaks at about 3 310 and 3 365 cm-1attributable to the different chemical environments of NH groups.1H NMR spectra of complex 1c show four NH resonance peaks at 7.92,7.90 3.71,and 3.56,respectively.The solid-state structures of 1a and 1b were determined by X-ray analysis.
As shown in Fig.1,X-ray analysis results show that 1a and 1b are isostructural,four o-aminobenzamido dianion ligands connect with two CpLn and two CpLn(HMPA)fragments via two different μ-η2∶η2-and μ3-η1∶η1∶η2-bonding modes,respectively.These bond parameters indicate that the amido moiety acts as both a bridging and side-on chelating group,in which the negative charge is delocalized over the OCN unit.Characteristically,both of bridging O and N atoms have two distinctive metal-oxygen (nitrogen)distances.One is between those expected for an Ln3+-O(N)single bond and an Ln3+←∶O(N)donor bond,while another falls in the range of the Ln3+←∶O(N)donating bond lengths for neutral oxygen or nitrogen donor ligands[28].This difference may be attributed to the chelating coordination effect caused by the amido ligand.
Scheme 4
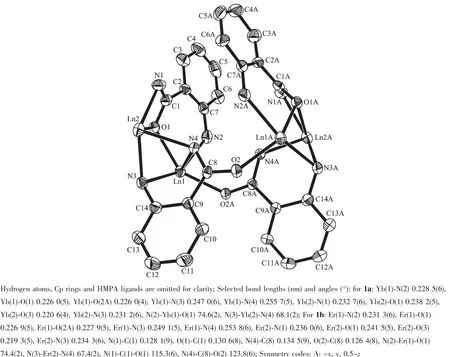
Fig.1 Thermal ellipsoid (30%)plot of complexes{CpLn(μ-η2∶η2-NHC6H4CONH)[μ3-η1∶η1∶η2-NHC6H4CONH]LnCp-(HMPA)}2 (Ln=Yb,1a;Er,1b)
2.2 Reaction of 1 with phenyl isocyanate
With dianionic o-aminobenzamido organolanthanides 1 in hand,we next explored their reactivity toward phenyl isocyanate.As expected,treatment of 1 with PhNCO (nPhNCO/n1=4)in toluene led to the occurrence of an unusual tandem addition/cyclization/amine elimination/ligand redistribution reaction,giving the dianionic quinazolyldiolate lanthanide complexes{Cp2Ln[μ3-η2∶η2∶η1-Quo]}3Ln(HMPA)2(Ln=Yb,2a;Er,2b;Y,2c)in moderate isolated yields (Scheme 5).The elimination product PhNH2was unambiguously identified by GC-MS,but attempts to isolate another metal-containing productwere unsuccessful.The metathesis reactions of isocyanates with organolanthanide complexes are rarely observed[20-21].This is the first example of the amido complex-mediated C=N bond cleavage ofisocyanates,and provides an effective method for the construction of quinazolyldiol skeleton[29].

Scheme 5

Fig.2 Thermal ellipsoid (30%)plot of complex{Cp2Yb[μ3-η2∶η2∶η1-Quo]}3Yb(HMPA)2 (2a)
Complexes 2a~2c are readily dissolved in THF but slightly soluble in toluene.They were characterized by elemental analysis and IR spectroscopy.The1H NMR spectra of 2c was also determined and showed that there is no N-H resonance absorption peak.The structure of 2a was identified by X-ray single-crystal diffraction analysis.As shown in Fig.2,2a is a tetranuclearstructure possessing three bridging dianionic quinazolinyldione ligands.2a has a three-fold rotation axis on the O-O alignment,and the three ytterbium and two HMPA oxygen atoms occupy an equatorial plane and an apical position of the hexahedral skeletal structure,respectively (Fig.2).The four Yb atoms locate in two different coordinate environments.Atoms Yb(2),Yb(2A)and Yb(2B)are coordinated by one chelating η2-N,O unit,one nonbridging oxygen atom and two η5-Cp groups,respectively,to form a distorted triagonal-bipyramidal geometry,while the central Yb(1)is coordinated by three η2-N,O units from different quinazolyldiolate ligands and two HMPA oxygen atoms to form a hexagonal bipyramidal geometry.The bond parameters indicate that the π-electrons of C=O and C=N double bonds are delocalized over the pyrimidinyldionate skeleton.The Yb-O distances are in normal range[5c].The rather long distances between ytterbium and N atoms may be attributed to both the rigidity of the quinazolyldiolate ring and the steric crowding[30].
2.3 Reactivity of 1 toward diisopropylcarbodiimide
To obtain additional insight into the mechanism and scope of the reaction,we further examined the behavior of 1 toward carbodiimide.In markedly contrast to isocyanate,treatment of 1 withiPrN=C=NiPr (nligand/n1=4)provided{Cp2Ln[μ-η1∶η1∶η2-iPrNC(NHiPr)NC6H4CONH]}3Ln(HMPA)3(Ln=Yb,3a;Er,3b;Y,3c)as the main metal-containing products,even with a prolonged heating at 110 ℃ (Scheme 6).In addition,3a~3c are inactive to excessiPrN=C=NiPr.These results indicate thatiPrN=C=NiPr is preferentially added to NH rather than CONH group.The coordinated CONH group is inert,and undergoes neither addition toiPrN=C=NiPr nor nucleophilic attack to the newly formed guanidinate ligand under the conditions involved.This difference might be attributed to the strong chelating interaction between CONH and metal,leading to the lower nucleophilicity compared with the NH group.
Crystallographic data for 3 show that there are two independent molecules (Fig.3),crystallizing in one asymmetrical unit.Itisnoteworthythatthe πelectrons of the C=N double bond of two guanidinate groups are delocalized over the N-C-N unit(N(2)-C(1)and N(4)-C(1),0.135(2)and 0.134(2)nm;C(49)-N(10)and C(49)-N(12),0.136(2)and 0.135(2)nm),whereas the bond parameters (N(6)-C(25)and N(8)-C(25)of a third guaninate group suggest a tendency toward CN double and single bonds(cf.mean values of 0.136 nm for C(sp2)-N and 0.129 nm for C(sp2)=N)[31].
In order to exclude the effect of HMPA,the HMPA-free reaction was examined.As shown in Scheme 6,the treatment of Cp3Ln with o-aminobenzamide followed by reacting withiPrN=C=NiPr gave{CpLn [μ-η1∶η2∶η2-NHCOC6H4NC (NHiPr)NiPr]}2(Ln=Yb,4a;Er,4b;Y,4c),indicating that the CONH group in 4 is also inert.Interestingly,4 can be transformed into 3 in the presence of HMPA,revealing an unusual HMPA-induced ligand redistribution of organolanthanides.
Crystals suitable for a single-crystal X-ray study were grown by slow cooling a hot toluene solution of 4.As shown in Fig.4,compounds 4a,4b and 4c are isostructural,and each of the lanthanide atoms is coordinated by one η5-C5H5ligand,one chelating η2-CONH with side donating coordination,one chelating guanidinate group and one bridging oxygen atom,indicating that only NHAr groups have combined withiPrN=C=NiPr.There are no informal bond lengths and anglescompared to otherguanidinate lanthanide complexes[27].

Scheme 6
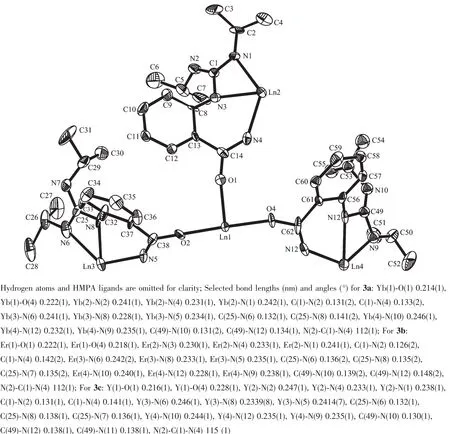
Fig.3 Thermal ellipsoid (30%)plot of complex{Cp2Ln[μ-η1∶η2∶η2-iPrNC(NHiPr)NC6H4CONH]}3Ln(HMPA)3(Ln=Yb,3a;Er,3b;Y,3c)

Scheme 7
Based on these experimental results,a possible reaction pathway for the formation of 2 is proposed in Scheme 7.Coordination insertion of PhNCO into the Ln-NHAr bond followed by a ligand redistribution give the intermediateⅡ,which places the CONH group and the resulting ureido into close proximity,as observed in the formation of 3.A sequential addition of the activated N-H to the C=N double bond leads to the occurrence of the cyclization[13].Reductive elimination of PhNH2generates the quinazolyldiolate ring.Obviously,the formation of a more stable aromatic quinazolyldiolate skeleton might contribute to the occurrence of the cyclization/amine-elimination reaction of the putative amido intermediate(Ⅲ).But attempts to isolate the intermediatesⅠandⅡhave been unsuccessful due to their thermodynamic instability.A tandem addition/cyclization/elimination reaction is observed in the treatment of 1 with PhNCO,whereas for 3 no subsequent reactions are observed even with a longer reaction time and higher temperature.It is possible that both the larger steric hindrance and the weaker electrophilic reactivity of guanidinate compared to ureido prevent the subsequent cyclization.

Fig.4 Thermal ellipsoid (30%)plot of complex{CpLn[μ-η1∶η2∶η2-OCNHC6H4NC(NHiPr)NiPr]}2 (Ln=Yb,4a;Er,4b;Y,4c)
3 Conclusions
In conclusion,three new tetranuclear lanthanocene derivatives incorporating the o-aminobenzamido dianion ligand,[CpLn(μ-η2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)LnCp-(HMPA)}2(Ln=Yb,Er,Y),have been synthesized by reaction of Cp3Ln with oaminobenzamide followed by crystallization in a mixed HMPA and toluene solvent.The first example of tandem reaction of two coordinated NH moieties with an isocyanate molecule resulting in the formation of a dianionic quinazolyldiolate fragmentisdescribed.Furthermore,these o-aminobenzamido complexes can also undergo the single NH addition to carbodiimide selectively.In addition,an unusual HMPA-induced ligand redistribution of organolanthanides is observed in these processes.The present results demonstrate that o-aminobenzamido organolanthanide complexes can be expected to have synthetic potential because they provide a reaction system in which the two coordinated NH moieties show different reactivity toward the same functionality.In this case,after the first reaction one surviving NH group could undergo cyclization across the first reaction product.On the other hand,this work provides an alternative route to carbonylation of organic substrates,which is very difficult to achieve by rare earth metal system due to the mismatch in the orbital energy of the hard rare earth metal ion and the soft CO ligand.The catalytic synthesis of heterocycles,which is based on such reactions,is undergoing.
[1] (a)Molander G A,Romero J A C.Chem.Rev.,2002,102(6):2161-2185(b)Muller T E,Hultzsch K C,Yus M.Chem.Rev.,2008,108(9):3795-3892(c)Bartoli G,Marcantoni E,Marcolini M,et al.Chem.Rev.,2010,110(10):6104-6143(d)Zeng X M.Chem.Rev.,2013,113(8):6864-6900
[2] (a)Evans W J,Davis B L.Chem.Rev.,2002,102(6):2119-2136(b)Zhou X G,Zhu M.J.Organomet.Chem.,2002,647(1/2):28-49(c)Hong S,Marks T J.Acc.Chem.Res.,2004,37(9):673-686(d)Zhang J,Zhou X G.Dalton Trans.,2011,40(38):9637-9648(e)Liu R T,Zhou X G.Chem.Commun.,2013,49(31):3171-3187
[3](a)Jeske G,Lauke H,Mauermann H,et al.J.Am.Chem.Soc.,1985,107(26):8091-8103(b)Cui D M,Nishiura M,Hou Z M.Angew.Chem.Int.Ed.,2005,44(6):959-962(c)Wang B L,Wang D,Cui D M,et al.Organometallics,2007,26(13):3167-3172(d)Kaneko H,Nagae H,Tsurugi H,et al.J.Am.Chem.Soc.,2011,133(49):19626-19629(e)Zhou S L,Wang H Y,Ping J,et al.Organometallics,2012,31(5):1696-1702(f)Hong L C,Lin W J,Zhou X G,et al.Chem.Commun.,2013,49(49):5589-5591
[4] (a)Li W B,Xue M Q,Xu F,et al.Dalton Trans.,2012,41(27):8252-8260(b)Basalov I V,Lyubov D M,Fukin G K,et al.Angew.Chem.Int.Ed.,2012,51(14):3444-3447(c)Sun J L,Berg D J,Twamley B.Organometallics,2008,27(4):683-690(d)Yang Y,Cui D M,Chen X S.Dalton Trans.,2010,39(16):3959-3967(e)Li X Q,Hong J Q,Zhou X G,et al.Organometallics,2010,29(20):4606-4610(f)Pi C F,Li X Q,Zhang L L,et al.Inorg.Chem.,2010,49(17):7632-7634(g)Zhang Z X,Bu X L,Zhang J,et al.Organometallics,2010,29(9):2111-2117(h)Lu E L,Chen Y F,Leng X B.Organometallics,2011,30(20):5433-5441(i)Casely I J,Ziller J W,Evans W J.Organometallics,2011,30(18):4873-4881
[5](a)Liu R T,Zhang C M,Zhu Z Y,et al.Chem.Eur.J.,2006,12(26):6940-6952(b)Li X Q,Liu R T,Zhang Z X,et al.Organometallics,2010,29(15):3298-3302(c)Chu J X,Lu E L,Liu Z X,et al.Angew.Chem.Int.Ed.,2011,50(33):7677-7680(d)Shao Y L,Zhang F J,Zhang J,et al.Angew.Chem.Int.Ed.,2016,55(38):11485-11489(e)Hong J Q,Li Z H,Chen Z N,et al.Dalton Trans.,2016,45(15):6641-6649(f)Huang S J,Shao Y L,Zhang L X,et al.Angew.Chem.Int.Ed.,2015,54(48):14452-14456(g)Luo Y,Teng H L,Nishiura M,et al.Angew.Chem.Int.Ed.,2017,56(31):9207-9210
[6] (a)Yasuda H,Yamamoto H,Yokota K,et al.J.Am.Chem.Soc.,1992,114(12):4908-4910(b)Giardello M A,Yamamoto Y,Brard L,et al.J.Am.Chem.Soc.,1995,117(11):3276-3277(c)Zhang W X,Nishiura M,Hou Z M.Angew.Chem.Int.Ed.,2008,47(50):9700-9703(d)Yang J Y,Shen H,Xie Z W.J.Organomet.Chem.,2015,798:204-208(e)Xu L,Zhai M K,Wang F,et al.Dalton Trans.,2016,45(43):17108-17112(f)Song G Y,Wang B L,Nishiura M,et al.Chem.Eur.J.,2015,21(23):8394-8398(g)Xu P F,Yao Y M,Xu X.Chem.Eur.J.,2017,23(6):1263-1267
[7] (a)Molander G A,Romero J A C.Chem.Rev.,2002,102(6):2161-2185(b)Konkol M,Kondracka M,Voth P,et al.Organometallics,2008,27(15):3774-3784(c)Ohashi M,Konkol M,Rosal D,et al.J.Am.Chem.Soc.,2008,130(22):6920-6921(d)Barros N,Eisenstein O,Maron L.Dalton Trans.,2010,39(44):10757-10767(e)Abinet E,Spaniol T P,Okuda J.Chem.Asian J.,2011,6(2):389-391
[8](a)Lauterwasser F,Hayes P G,Piers W E,et al.Adv.Synth.Catal.,2011,353(8):1384-1390(b)Zhang Y Y,Yao W,Li H,et al.Organometallics,2012,31(13):4670-4679(c)Trambitas A G,Melcher D,Hartenstein L,et al.Inorg.Chem.,2012,51(12):6753-6761(d)Reznichenko A L,Hultzsch K C.Organometallics,2013,32(5):1394-1408(e)Huang S J,Shao Y L,Zhang L X,et al.Angew.Chem.Int.Ed.,2015,54(48):14452-14456(f)Hong S,Marks T J.Acc.Chem.Res.,2004,37(9):673-686
[9] (a)Yu X H,Seo S Y,Marks T J.J.Am.Chem.Soc.,2007,129(23):7244-7245(b)Seo S Y,Yu X H,Marks T J.J.Am.Chem.Soc.,2009,131(1):263-276
[10](a)Weiss C J,Marks T J.Dalton Trans.,2010,39:6576-6588(b)Weiss C J,Wobser S D,Marks T J.Organometallics,2010,29(23):6308-6320
[11](a)Douglass M R,Stern C L,Marks T J.J.Am.Chem.Soc.,2001,123(42):10221-10238(b)Takaki K,Koshoji G,Komeyama K,et al.J.Org.Chem.,2003,68(17):6554-6565(c)Kawaoka A M,Marks T J.J.Am.Chem.Soc.,2004,126(40):12764-12765(d)Hu H F,Cui C M.Organometallics,2012,31(3):1208-1211(e)Behrle A C,Schmidt J A R.Organometallics,2013,32(5):1141-1149
[12](a)Hung S C,Wen Y F,Chang J W,et al.J.Org.Chem.,2002,67(4):1308-1313(b)SchumannH,HeimA,DemtschukJ,etal.Organometallics,2003,22(1):118-128(c)Yuan Y Y,Wang X F,Li Y X,et al.Organometallics,2011,30(16):4330-4341
[13](a)Guan B T,Hou Z M.J.Am.Chem.Soc.,2011,133(45):18086-18089(b)Oyamada J,Hou Z M.Angew.Chem.Int.Ed.,2012,51:12828-12832(c)Guan B T,Wang B L,Nishiura M,et al.Angew.Chem.Int.Ed.,2013,52:4418-4421(d)Shi X C,Nishiura M,Hou Z M.J.Am.Chem.Soc.,2016,138:61476150(e)Arnold P L,McMullon M W,Rieb J,et al.Angew.Chem.Int.Ed.,2015,55:82-100
[14](a)Monsaert S,Vila A L,Drozdzak R,et al.Chem.Soc.Rev.,2009,38:3360-3372(b)Leitao E M,van der Eide E F,Romero P E,et al.J.Am.Chem.Soc.,2010,132(8):2784-2794(c)Woodward C P,Spiccia N D,Jackson W R,et al.Chem.Commun.,2011,47:779-781
[15](a)Shima T,Hou Z M.J.Am.Chem.Soc.,2006,128 (25):8124-8125(b)Zhou J L,Chu J X,Zhang Y Y,et al.Angew.Chem.Int.Ed.,2013,52(15):4243-4246
[16](a)Zhang W X,Wang Z,Nishiura M,et al.J.Am.Chem.Soc.,2011,133(15):5712-5715(b)Hong J Q,Zhang L X,Yu X Y,et al.Chem.Eur.J.,2011,17(7):2130-2137(c)Hong J Q,Zhang L X,Wang K,et al.Chem.Eur.J.,2013,19(24):7865-7873
[17](a)Masui H,Fuse S,Takahashi T.Org.Lett.,2012,14(16):4090-4093(b)Campbell M J,Toste F D.Chem.Sci.,2011,2(7):1369-1378(c)Attanasi O A,de Crescentini L,Favi G S.et al.Org.Lett.,2011,13(3):353-355(d)Groenendaal B,Vugts D J,Schmitz R F,et al.J.Org.Chem.,2008,73(2):719-722(e)Church T L,Byrne C M,Lobkovsky E B,et al.J.Am.Chem.Soc.,2007,129(26):8156-8162
[18](a)Braunstein P,Nobel D.Chem.Rev.,1989,89 (8):1927-1945(b)Kuninobu Y,Tokunaga Y,Kawata A,et al.J.Am.Chem.Soc.,2006,128(1):202-209(c)Paul F,Moulin S,Piechaczyk O,et al.J.Am.Chem.Soc.,2007,129(23):7294-7304(d)Zhu X C,Fan J X,Wu Y J,et al.Organometallics,2009,28(13):3882-3888(e)Sharpe H R,Geer A M,Williams H E L,et al.Chem.Commun.,2017,53(5):937-940
[19](a)Evans W J,Forrestal K J,Ziller J W.J.Am.Chem.Soc.,1998,120(36):9273-9282(b)Shen Q,Li H R,Yao C S,et al.Organometallics,2001,20(14):3070-3073(c)Shen Q,Yao Y M.J.Organomet.Chem.,2002,647(1/2):180-189(d)Han Y N,Zhang J,Han F Y,et al.Organometallics,2009,28(13):3916-3921(e)Yi W Y,Zhang J,Li M,et al.Inorg.Chem.,2011,50(22):11813-11824
[20](a)Zhang J,Ma L P,Cai R F,et al.Organometallics,2005,24(4):738-742(b)Du Z,Zhou H,Yao H,et al.Chem.Commun.,2011,47(12):3595-3597
[21]Tardif O,Hashizume D,Hou Z M.J.Am.Chem.Soc.,2004,126(26):8080-8081
[22]Qian C T,Ye C Q,Lu H Z,et al.J.Organomet.Chem.,1983,247(2):161-170
[23]SAINTPlus,Data Reduction and Correction Program Ver.6.02a,Bruker AXS,Madison,WI,2000.
[24]Sheldrick G M.SADABS,A Program for Empirical Absorption Correction,University of Göttingen,Germany,1998.
[25]Sheldrick G M.SHELXL-97,Program for the Refinement of Crystal Structures,University of Göttingen,Germany,1997.[26](a)Sun Y,Zhang Z X,Wang X,et al.Organometallics,2009,28(21):6320-6330(b)Sun Y,Zhang Z X,Wang X,et al.Dalton Trans.,2010,39(1):221-226(c)Zhang J,Han Y N,Han F Y,et al.Inorg.Chem.,2008,47(13):5552-5554
[27](a)Pi C F,Liu R T,Zheng P Z,et al.Inorg.Chem.,2007,46(13):5252-5259(b)Pi C F,Zhu Z Y,Weng L H,et al.Chem.Commun.,2007(21):2190-2192(c)Pi C F,Zhang Z X,Pang Z,et al.Organometallics,2007,26(8):1934-1946
[28](a)Evans W J,Ulibarri T A,Chamberlain L R,et al.Organometallics,1990,9(7):2124-2130(b)Evans W J,Foster S E.J.Organomet.Chem.,1992,433(1/2):79-94(c)Venugopal A,Kamps I,Bojer D,et al.Dalton Trans.,2009(29):5755-5765
[29](a)Willis M C,Snell R H,Fletcher A J,et al.Org.Lett.,2006,8(22):5089-5091(b)Vorbrueggen H,Krolikiewicz K.Tetrahedron,1994,50(22):6549-6558(c)Li J R,Chen X A,Shi D X,et al.Org.Lett.,2009,11(6):1193-1196(d)Patil Y P,Tambade P J,Deshmukh K M,et al.Catal.Today,2009,148(3/4):355-360
[30]Zhou X G,Huang Z E,Cai R F,et al.J.Organomet.Chem.,1998,563(1/2):101-112
[31]Allen F H,Kennard O,Watson D G,et al.J.Chem.Soc.,Perkin Trans.,1987(12):S1-S19
Syntheses,Cycloaminocarbonylation and Amidination of Rare Earth o-Aminobenzamido Dianion Complexes Bearing Cyclopentadienyl Co-ligand
SUN Yan1LIU Rui-Ting1WENG Lin-Hong1ZHOU Xi-Geng*,1,2
(1Department of Chemistry,Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials,Fudan University,Shanghai 200433,China)
(2State Key Laboratory of Organometallic Chemistry,Shanghai 200032,China)
Treatment of Cp3Ln with o-aminobenzamide followed by crystallization in a HMPA and toluene mixture affords the tetranuclear organolanthanide complexes [CpLn(μ-η2∶η2-NHC6H4CONH)(μ3-η1∶η1∶η2-NHC6H4CONH)LnCp(HMPA)}2(Ln=Yb,1a;Er,1b;Y,1c).Reaction of 1 with PhNCO (nPhNCO/n1=4)in toluene gives the dianionic quinazolyldiolate (Quo)complexes [Cp2Ln(μ3-η2∶η2∶η1-Quo)]3Ln(HMPA)2(Ln=Yb,2a;Er,2b;Y,2c),indicating that one isocyanate molecule can undergo the tandem reaction with both NH and CONH of 1 to construct a quinazoly-ldiolate skeleton,companying with the elimination of PhNH2.However,1a~1c react withiPrN=C=NiPr under the same conditions to give only the single ArNH addition products{Cp2Ln [μ-η1∶η1∶η2-iPrNC(NHiPr)NC6H4CONH]}3Ln (HMPA)3(Ln=Yb,3a;Er,3b;Y,3c).Furthermore,treatment of Cp3Ln with o-aminobenzamide followed by reacting withiPrN=C=NiPr gave{CpLn[μ-η1∶η2∶η2-NHCOC6H4NC(NHiPr)NiPr]}2(Ln=Yb,4a;Er,4b;Y,4c).Noticeably,HMPA could induce the transformation of 4 into 3 by a ligand redistribution.CCDC:951003,1a;951104,1b;951102,2a;951004,3a;951106,3b;951005,3c;951103,4a;951107,4b;951105,4c.
lanthanide complexes;cycloaminocarbonylation;isocyanate;o-aminobenzamide;addition
O614.346;O614.344;O614.32+2
A
1001-4861(2017)11-2124-15
10.11862/CJIC.2017.255
2017-09-01。收修改稿日期:2017-09-22。
国家自然科学基金(No.21372047,21572034)资助项目。
*通信联系人。E-mail:xgzhou@fudan.edu.cn
