二维半导体材料与器件
——从传统二维光电材料到石墨炔
2017-11-13黄彦民袁明鉴李玉良
黄彦民 袁明鉴*, 李玉良
二维半导体材料与器件
——从传统二维光电材料到石墨炔
黄彦民1袁明鉴*,1李玉良2
(1南开大学化学学院,南开大学先进能源材料化学教育部重点实验室,天津, 300071)
(2中国科学院化学研究所有机固体重点实验室,北京分子科学国家实验室,北京 100080)
近年来,二维半导体材料由于其独特的材料结构和电子输运特性得到了科学界的广泛关注,被应用于光电器件、催化和生物传感器等领域。本文系统概述了传统二维材料以及新兴二维材料石墨炔的发现和发展历程。重点聚焦在二维材料在光探测器领域中的应用,探讨了不同二维材料体系及器件结构对光探测器性能的影响;并详细介绍了新兴二维材料——石墨炔,及其合成和应用。展望了传统二维材料及石墨炔在光电转换器件应用中所面临的机遇和挑战。
二维半导体材料;石墨炔;光电转换材料与器件
0 引 言
科学家们一直对低维半导体材料的研究有着浓厚的兴趣,继零维富勒烯和一维单壁碳纳米管被发现后[1-2],又将研究兴趣转到了二维材料上。80多年前,著名的物理学家Landau等就预测准二维晶体材料因其热力学状态不稳定而不能单独稳定存在;随后Mermin和Wagner又进一步指出,二维材料表面的弯曲褶皱会破坏其有序性,与Landau预测结果相一致[3]。然而,这些问题并不能阻止科学家们对二维材料可控制备方法的探索。1962年Boehm等[4]就已经制备得到了少层的氧化石墨烯,并且此后科学家们不断地报道了类似的发现,但是单层石墨烯能否稳定存在一直都没有定论。直到2004年,英国的两位科学家Geim和Novoselov开创了二维材料可控制备的先河[5],他们利用胶带反复剥离的方法在绝缘衬底上得到了单层的石墨烯,打破了之前的预言并填补了单原子层二维材料领域中的空白。石墨烯因具有二维量子限域效应被广泛应用于纳米光电等领域[6-7]。除石墨烯之外,Geim还发现许多类石墨烯材料,尤其是过渡金属硫族化合物(Transition Metal Dichalco-genides,TMDs)也能通过剥离的方法得到稳定存在的单层膜[8]。这类物质大都具有X-M-X的层状结构,金属原子M所在的六方结构层以三明治夹心的方式被X层所包围。进一步研究发现单层的TMDs所表现出来的特性与其对应的体材料有很大的不同[9-10]。 例如,当二硫化钼(MoS2)从体材料变为单层时,其由间接带隙半导体(1.2 eV)转变为直接带隙半导体(1.8 eV)[11]。此外Ⅲ~Ⅳ硫族化合物(GaS、GaSe、InSe、SnS、SnS2)、黑磷(Black Phosphorus,BP)、过渡金属氧化物、拓扑绝缘体以及新兴二维材料石墨炔等一系列材料也被广泛开发并应用于光电器件、催化、生物传感器等领域[12-15]。
本文回顾了传统二维半导体材料在光探测器件中的应用,并探讨了不同二维材料体系及器件结构对光探测器性能的影响;详细介绍了新兴二维材料石墨炔,及其合成和应用,同时展望了二维半导体材料在光电转换领域所面临的机遇及挑战。
1 二维材料的结构、特性及制备
1.1 二维材料的结构及性质
2004年单层石墨烯的发现开创了二维材料研究的先河,然而到目前为止科学界还没有给出“二维材料”非常明确的定义[16]。目前,科学家们对一种材料是否可以定义为二维材料有以下一些主流的判断标准[17]:(1)量子限域效应:电子被限域在超薄的二维层状结构中,尤其是单原子层厚度(即在垂直方向上超薄),没有层间作用。(2)在二维平面上生长,具有较大的横向尺寸。(3)原子排列有序,呈现一定的晶体性质及各向异性。
石墨烯由sp2杂化的碳原子组成,并由六方环状结构紧密排列成蜂窝状 (如图1a所示);无论是ABAB还是ABC的堆叠方式,石墨烯层与层之间弱的范德华作用力留给了科学家们很大的想象空间。尽管不同材料在晶体结构上有所不同,但是它们大都被称为层状材料,这是因为它们与石墨烯有着共同的特点:在层状平面内由强的共价键将原子连接起来并按照一定规则排列;而在层与层之间靠的是弱的范德华作用力相互堆叠,正是因为这种层间弱范德华力堆叠的特性使科学家们得以通过简单的剥离方法对其进行初步研究[5]。
石墨烯的量子限域效应和特殊的能带结构,决定了石墨烯电子的无质量狄拉克费米子属性[18](如图1b所示),这种独特的电子特性使石墨烯成为了量子霍尔效应、克莱因佯谬等凝聚态物理现象的绝佳研究材料[16];更重要的是在高质量的单晶石墨烯中,其载流子可以不散射地传输数十微米[20-22]。正是这种超长的载流子扩散长度和超高的导电性使石墨烯在电子和光电器件中发挥了极大作用。另一种被广泛研究的二维半导体材料是MoS2,图1(c~d)为MoS2的材料能带结构和禁带宽度的变化。值得注意的是当MoS2从体材料逐渐变为单层时,其由间接带隙半导体(禁带宽度约为1.3 eV)转变为直接带隙半导体[11](禁带宽度约为 1.8 eV);相比较于没有带隙的石墨烯,以MoS2为首的其它层状二维半导体材料似乎更适合作为光电器件的沟道材料[16]。
1.2 二维材料的制备
之前我们已经提到了超薄二维材料的层状结构,正是由于层状结构中这种弱的层间范德华作用力,二维半导体材料可以通过各种各样的自上而下的剥离方法(机械剥离、液相剥离、锂离子插层剥离、阳离子交换剥离)实现可控制备。其它材料由于晶体结构在三维方向上全部形成共价键或者离子键,不具备弱的层间范德华作用力难以剥离。各种各样的二维半导体材料的剥离方法大都依赖于外力的驱使来打破层间范德华作用力,从而得到少层甚至单层的半导体材料[23-24]。除此之外,自下而上的合成方法也不断被报道,这类方法主要依靠控制反应条件使具有不同理化性质的前驱体反应而生成层状材料,其中发展最为成熟的是化学气相沉积法和湿法化学合 成法[16]。

图1 (a)碳六边形结构;(b)石墨烯的能带结构[18];(c)MoS2布里渊区示意图[19];(d)体相 TMDs 和单层 MoS2的能带结构[11]Fig.1 (a)Structure of hexagonal carbon;(b)Band structure of graphene[18];(c)Brillouin zone of TMDs[19];(d)Band alignment of single and bulk MoS2[11]
(1)机械剥离法:这是最早被报道制备单层二维材料的方法。简单来说就是将块体层状石墨在胶带之间反复粘附,使层状晶体逐渐减薄。当到达一定程度之后就可将胶带贴在所需衬底上,利用加热、按压等手段将超薄石墨烯纳米片转移至衬底上。由于衬底的反光作用,得到的石墨烯纳米片可以在光学显微镜下被明显观察到。但通过机械剥离方法所得到的二维材料厚度和形貌都不均匀,尺寸较小,且少数层膜产率很低。尽管如此,这种廉价易得的方法仍是目 前研究 二维半 导体材 料的重 要制备 方法[5,12,16]。
(2)液相剥离法:液相剥离法也是一种简便易得的制备超薄二维材料的方法。将层状体材料分散在N,N-二甲基甲酰胺(DMF)或 N-甲基吡咯烷酮(NMP)等溶剂中,在超声波作用下就可以在不破坏平面内共价键的前提下打破弱的层间范德华作用力,得到超薄二维纳米片的悬浊液;进而通过离心分离的方法根据转速的不同,得到具有不同层数分布二维材料的悬浊液。这种方法的剥离产率跟溶剂和材料之间的表面能有很大关系:降低体系的能量有利于材料的剥离和纳米片的分散。此外,由于表面活性剂有利于体系的稳定,使用表面活性剂可以更有效地对二维材料进行剥离。目前利用该方法已经得到了如石墨烯、TMDs、金属氧化物、BP等超薄的纳米片[25-30]。虽然该方法有利于大规模制备超薄纳米片,但制备得到的横向尺寸通常不大,一般不超过5 μm,并且对材料性能有害的有机溶剂和表面活性剂会吸附在材料表面难以去除。
(3)离子插层/电化学剥离法:该方法将离子插入体相材料层间以减弱层间范德华作用力,进一步通过超声的方法使超薄纳米片之间相互分离。将体相层状材料和铜箔分别作为电化学池的阴极和阳极,通过离子插层作用达到剥离效果[31]。这种方法制备得到的单层二维材料产率非常高,如MoS2的单层产率能达90%以上;同时离子插层也会给材料带来一些奇特的性能,例如TMDs经过插层之后会由原来的半导体相转变为金属相,有利于降低其接触电阻,从而有利于其在电催化等领域的应用[32-33]。
(4)化学气相沉积法:目前化学气相沉积法是制备大尺寸、厚度可调控、高质量单晶的较好方法。但目前该方法也存在一定问题,一般需要具有较高活性的催化基底,在后期使用中需要转移,此外高温制备所得到的单晶存在较多缺陷。因此发展无转移、低温条件下制备超大尺寸二维单晶材料的方法亟待展 开[34-38]。
(5)液相合成法:该方法通常用来合成特定的目标结构(如0D-2D复合结构),通过表面活性剂和反应物浓度等方式来控制产物的尺寸、厚度等形貌。该方法目前存在的问题主要是单层和较大尺寸的产物仍 然难以 制得[16,39]。
2 二维材料在光探测器中的应用
目前以二维材料为核心的高性能光探测器的研究主要集中在特定纳米结构及器件结构设计上,选择具有优良性能的材料是制备具有不同光谱响应范围和光响应性能光探测器的基础。本章首先回顾了不同二维材料光探测器的本征性能,其次,探讨了不同复合结构对二维材料光探测性能的影响。
2.1 光探测器的基本原理及评价参数
最简单的光探测器系统为两电极的光导器件,如图2a中Ⅰ及2b所示,黑暗环境中,在外加电场的作用下载流子从电极注入并通过沟道材料。通常由于沟道材料本身的电阻值较大,其暗电流一般较小;然而,在光照条件下,半导体材料能够有效地吸收光子从而产生电子空穴对,在外电场的作用下,电子-空穴分离并分别注入到相应的电极中,从而显著增加了材料中的载流子浓度,使得其光电流显著增加且高过暗电流数个数量级。为了定义半导体材料这种优异的光响应性能,科学家们引入了如下参数对其性能进行评价。
光响应度(R):

Iλ:净的光电流(器件受光照时的电流大小减去黑暗时的电流大小)
P:单位面积下的光照强度
S:器件在沟道中的有效面积
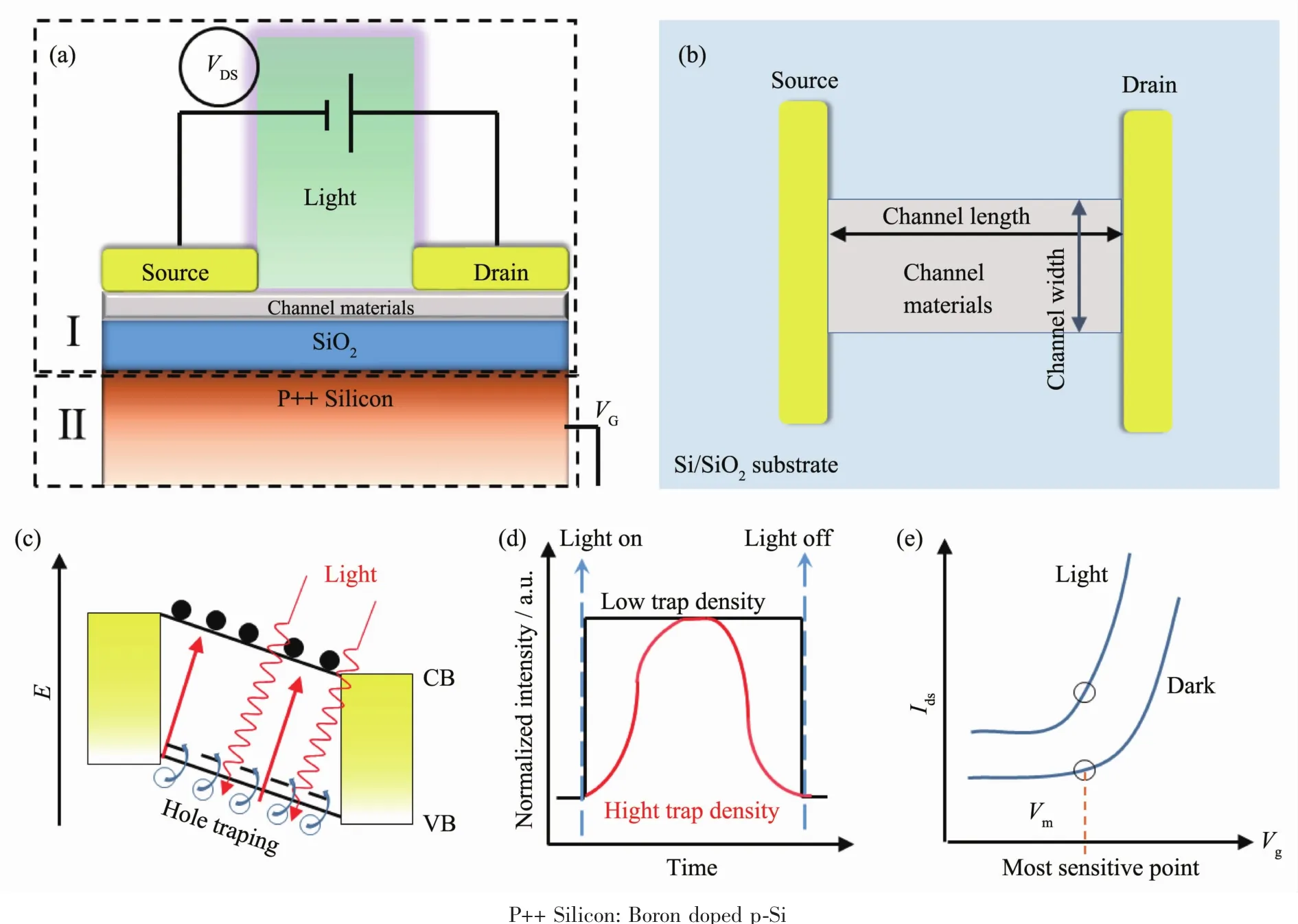
图2 (a,b)光探测器结构图;(c)光探测器响应机理;(d)势阱和载流子寿命对响应时间的影响;(e)光晶体管的最灵敏探测栅压Fig.2 (a,b)Scheme of 2D photodetector;(c)Mechanism of photodetector;(d)Effect of trap and carrier life to response time;(e)Most sensitive point in phototransistor
光探测器在工作过程中最为基础的过程就是将吸收的光子转换为电子-空穴对,而光响应度(R)就是用来衡量材料在单位光照强度下输出电流大小的能力。器件的光响应能力的大小跟入射光波长、入射光强、外加电场等因素相关。
归一化探测率(D*):

R:光响应度;
A:器件的有效面积;
e:电子电荷;
Id:暗电流大小;
归一化探测率(D*)则用来描述光探测器的灵敏度。该参数包含了2个重要的参数:光响应度和噪音平台的水平(与暗电流的大小相关)。光电流和暗电流的比值越大器件探测到光强度的变化情况就越灵敏。
外量子效率(EQE):

h:普朗克常数
R:光响应度
e:电荷电量
λ:入射光波长
外量子效率主要用来描述器件收集到的电子数与入射光子数之比。
光增益(G):
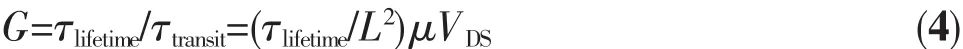
τlifetime:载流子的寿命
τtransit:载流子流经沟道所需要的时间
L:沟道长
μ:迁移率
VDS:漏源电压
在绝大多数光电探测器中,以n型材料为例(如图2c所示),沟道中的空穴会陷入势阱,而电子则可以在沟道中自由移动且不被复合,如果被捕获空穴的时间(载流子寿命)大于自由载流子(电子)流过沟道的时间就会产生光增益。如果一种材料具有高的迁移率和长的载流子寿命,那么它将会获得极高的光增益,使其外量子效率远远超过100%。
尽管高的光增益能使的器件的光响应度和归一化探测率等性能大大增加。但是长的载流子寿命也会使器件的响应时间大大增加(如图2(c,d)所示)。在光照条件下,电子从价带激发到导带,此时的电子容易和空穴复合,产生的光电流很小,当产生的空穴被浅能级的势阱捕获后,由于长的载流子寿命所带来的高的光增益使得器件获得较大的光电流,但填充过程使得器件的响应时间有所增加。同样在光场切换至黑场状态的瞬间,由于空穴不能马上从势阱中脱离与电子复合,材料中仍然保持较高的载流子浓度,随着空穴从浅能级势阱中逃离,电子空穴对的复合,光电流逐渐减小到暗电流时的水平。因此器件难以同时获得高的响应度和快的响应速率,其响应时间和响应速率的平衡是科学家们亟待解决的难题。
为了进一步优化器件的探测性能,光敏半导体材料通常被设计成光电晶体管的结构,如图2a中Ⅱ所示。通过调节栅极电压的大小,可以有效地调节材料的能带位置和沟道的导电特性,从而获得最佳探测条件。在最佳感应栅压下,器件能获得较大的净光电流,同时还能保证器件有较大的归一化探测率(如图 2f所示)。
2.2 本征光探测器
石墨烯材料具有优异的半导体性能如高的载流子迁移率、宽的光谱响应范围,但是由于没有带隙这一天然缺陷,导致其响应度(~0.01 A·W-1)和光增益效应(外量子效率:0.1%~0.2%)极低,因此不适合作为光探测器[40-41]。Zhang等通过对器件结构的重新设计,得到了具有高响应度(8.61 A·W-1)和高光增益的纯石墨烯光探测器[42]。 如图 3(a~c)所示,在将单层石墨烯制备成了标准光电晶体管之后,继续在石墨烯的表面覆盖一层厚度不均匀的金属钛。由于金属层的厚度不同,随后的刻蚀会将石墨烯的表面刻出许多量子点孔洞 (GDQ Array),从而打开石墨烯的带隙,使其在一个比较宽的范围内都有比较高的光响应。但是刻蚀剂会破坏石墨烯表面的结构产生大量的缺陷,虽然其光响应度得到了显著提高,但响应时间明显增加。
近期研究结果表明,具有直接带隙的其它超薄层状材料相比较于本征石墨烯在光探测器领域更具应用价值。 以 GaS 为例(图 3(d~f),在从体材料向二维材料转变的过程中其禁带宽度由2.59 eV逐渐变化为3.05 eV。由于带隙较宽,它非常适合作为紫外光探测器。基于高结晶度GaS纳米片的光探测器已经在刚性和柔性基底上得到了展现,具有较高的光响应度(19.6 A·W-1)和外量子效率(9 374%)。 进一步的理论计算表明随着层数的减少,GaS在价带顶的有效质量减少会促进其载流子迁移率增加,保证了GaS具有很高的光增益效果[43]。
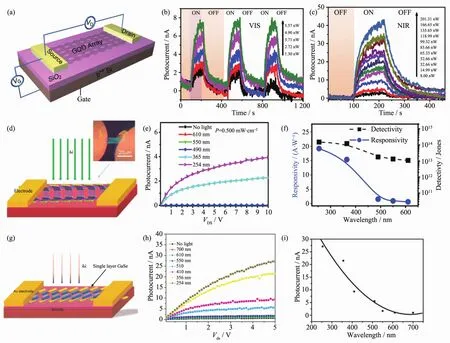
图3 (a~c)量子点图案化石墨烯光探测器[42];(d~f)GaS 光探测器[43];(g~i)GaSe 光探测器[44]Fig.3 (a~c)Quantum dots patterned graphene photodetector[42];(d~f)GaS photodetector[43];(g~i)GaSe photodetector[44]
另一种镓族材料GaSe,其间接带隙宽度(2.11 eV)与直接带隙非常接近(差值为25 meV)。尽管其在可见光区有一定的吸收,但是在可见光区域(波长大于550 nm)GaSe器件的光电流响应大为减小,如图3(g~i)所示。因此,尽管 GaSe 带宽较窄却只适宜作为紫外波段光探测器使用。在波长254 nm的紫外光激发下,其光响应度和外量子效率分别可达2.8 A·W-1和 1 367%[44]。
获得高性能可见光探测器的核心是探索具有更窄禁带宽度的二维半导体材料。MoS2具有 较窄的禁带宽度 (约1.8 eV)和较高的载流子迁移率(~70 cm2·V-1·s-1),并且其在 400~680 nm 的可见光区域具有较快、较高的光响应性能[45],响应时间约为50 ms。图 4(a~c)为单层 MoS2光电器件示意图及其光电性能表征。Oriol等[46]发现在低的激发光强度 (~24 μW·cm-2)下,MoS2器件的光响应度能达到超高的880 A·W-1。由于体相MoS2是一种间接带隙半导体(禁带宽度约为 1.3 eV),Woong 等[47]试图通过调控其厚度来调控MoS2的光响应范围。由于间接带隙的跃迁禁阻,其光探测器在可见光区的光响应度和归一化探测率在较低的光照强度50 mW·cm-2下,可达120 mA·W-1和1011Jones;但在850 nm红外光下,需要一个超高的光照强度(2.3 W·cm-2),才有极低的光响应度 (0.09 mA·W-1)和归一化探测率(~107Jones)。由此可见,窄带隙的间接带隙半导体材料MoS2并不适合用于红外光区域的光探测器件(图4(d~f)。
另一与MoS2具有相似禁带宽度的二维材料是GaTe(禁带宽度约为1.7 eV)。值得注意的是,GaTe材料在从体相变为单层的过程中其带隙类型并没有发生明显的转变,均为直接带隙半导体,这就给GaTe材料带来了极大的可塑性[48]。当前少数层的GaTe纳米片光探测器的光响应度已经达到了274.4 A·W-1,归一化探测率在1011~1012Jones之间,表现出了优异的光响应性能[49]。而多层的GaTe光响应度更是达到了104A·W-1,并且具有很快地响应速度6 ms。上述指标较其它二维材料体系(如MoS2、石墨烯等)提升了很多,如图 4(g~i)所示。
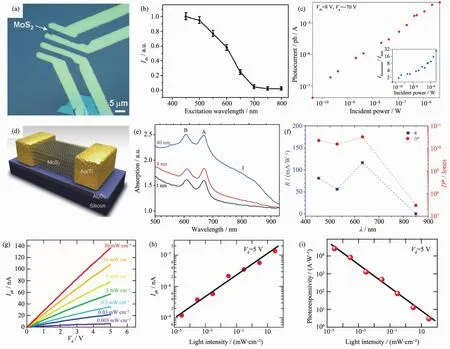
图4 (a~c)薄层 MoS2光探测器[45-46];(d~f)体相 MoS2光探测器[47];(g~i)GaTe 光探测器[48]Fig.4 (a~c)Ultratin MoS2photodetector[45-46];(d~f)Bulk MoS2photodetector[47];(g~i)GaTe photodetector[48]
为了进一步开发二维半导体材料在红外光区域的光探测应用,科学家们开始研究更窄带隙的二维材料,InSe即为其中之一。单层的InSe材料为直接带隙半导体,其禁带宽度约为1.3 eV,较间接带隙的体相InSe低约200 meV,因而是一种具有很好红外光响应的二维材料[50-53]。但是通常单层材料的吸光系数不高,导致其光响应性能较差。科学家们发现随着材料厚度的增加,其吸光系数存在一个极大值,而该厚度条件下的材料理论上是其作为光探测器的最佳厚度。Feng等[54]发现厚度约为30~40 nm厚的InSe在栅极电压为40 V,850 nm红外光条件下具有很高的光响应度 (~3×103A·W-1), 归一化探测率可达1012Jones,响应和衰减时间分别为5和8 ms,因此,该厚度条件下的InSe可以作为一种较为理想的红外光探测材料,如图 5(a,b)所示。
近年来科学家们发现二维材料中的BP是最有潜力成为高响应度、快速、宽光谱响应光探测器[56-59]。理论计算所得到的BP的禁带宽度在1~2 eV之间,通过材料厚度的调控,其禁带宽度可以在0.3~2 eV范围内实现调控。单层BP通过荧光光谱计算得到的禁带宽度约为1.32 eV。相较于MoS2、GaS、InSe等材料,BP 的迁移率非常高(~10 000 cm2·V-1·s-1)。在早期基于BP光探测器件的研究中[55],640 nm光激发下其光响应度可达4.8 mA·W-1,虽然响应度较MoS2材料低3~5个量级,但截止响应波长为997 nm,并且具有很快地响应速度(~1 ms),如图 5(c,d)所示。Wu等[60]还发现,在紫外光条件下,BP的光响应度可达~9×104A·W-1,归一化探测率也极高,约为3×1013Jones。但是BP在空气中难以稳定存在极大限制了它的应用。
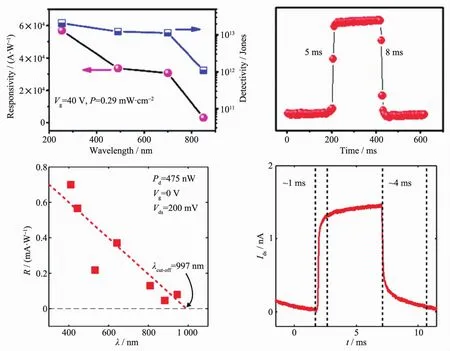
图5 (a,b)InSe 光探测器[54];(c,d)BP 光探测器[55]Fig.5 (a,b)InSe photodetector[54];(c,d)BP photodetector[55]
2.3 复合体系光探测器
上述本征光探测器由于受到材料本身特性的限制,光响应特性不能满足实际使用需求,因此科学家们开发了新的复合材料结构体系。由于不同组分材料在性能上可以相互弥补,复合结构光探测器在光谱响应范围、光响应度、迁移率、归一化探测率等性能参数上有显著改善。相较于单一的材料,复合结构光探测器的光响应度一般也能提高10倍以上,有的甚至能提高1 000倍以上,这种高的响应度大大超过了传统硅基材料,有巨大的应用前景。其结构主要分为两类:一种是电流流经闭合回路时需要经过不同类型的材料,即异质结;另一种则是以一种材料作为沟道导电基体,利用敏化的方式使其光电性能提高。
2.3.1 自驱动二维异质结光探测器
大多数二维半导体材料构成的异质结都依靠范德华力形成相互接触。这种范德华作用形成的材料间隙通常很小,数量级在~10-1nm。在大约1 V的内建电场控制下,电子和空穴能够发生快速转移,并分别向两极移动 (即使在没有外加电场的条件下也具有光响应特性)。这种光伏效应能让其具有优异的光电 探 测 性 能[61]。
图6(a~c)为 n-MoS2/p-WSe2异质结。 n-MoS2/p-WSe2具有良好的栅极调控光伏效应,从图中可以看出,当漏源电压为0 V时,器件的最大光响应度为2 mA·W-1;进一步的荧光光谱表明,相较于独立的纳米片,复合结构的MoS2和WSe2的荧光猝灭率分别为81%和98%,这种强烈的荧光猝灭效应是电子-空穴对有效分离所导致的,这也为我们研究异质结光电器件奠定了基础[62]。
此后,科学家们研究了很多不同组合的二维异质结,并对其性能进行了优化。Pezeshki等[63]制备得到了 MoS2/MoTe2异质结,如图 6(d~f)所示。 在 5 V 偏压下整流比为~1×103,尽管MoTe2的迁移率不高(0.2~40 cm2·V-1·s-1), 但其器件的响应速度依然很快,约为25 ms。在没有外加电场800 nm红外光激发下,其光响应度可达38 mA·W-1。与此同时,Zhang等[64]报道了MoS2/MoTe2异质结光探测器,他们利用开尔文探针显微镜、密度泛函理论及荧光光谱详细研究了MoS2/MoTe2所形成的type-Ⅱ型异质结构的能带位置及电荷分离特性。由于MoTe2的价带和MoS2的导带之间的层间耦合作用 (层间带宽:0.66 eV),使得MoS2/MoTe2光探测器在1 550 nm光激发下仍具有较大的光响应(图 6(g~i)。
此外,最具有潜在应用价值的BP异质结构光探测器也被报道。Deng等[65]利用p型掺杂的BP和n型掺杂的MoS2制备得到了p-n光电晶体管,如图7(a,b)所示。器件表现为明显的n型特性,并且栅极可调。在负的栅压下,器件表现出较高的整流比(~105)。并且在栅压为-40 V的条件下,其光电流/暗电流的比值可达3×103,较MoS2高出100倍以上。尽管由于负的栅压下器件具有很大的电阻使光电流大为减小,但此时对暗电流的压制作用更为明显。在正的栅压偏置下,其光响应度可达3.54 A·W-1,比单独的BP(~4.8 mA·W-1)高 700 倍以上。 Ye 等[66]进一步研究了BP/MoS2在近红外光条件下的光电响应。在绿光(532 nm)照射下,其光响应度可以达到22.3 A·W-1,即使在 1 550 nm 的近红外光照条件下(如图 7(c,d),该器件仍能拥有较强的光响应度 (153.4 mA·W-1)和归一化探测率(~2×109Jones)。此外该器件还具有超快的响应速度,响应时间和恢复时间分别为15和70 μs。相较于最近报道的石墨烯异质结器件,其器件响应还要快数个数量级。
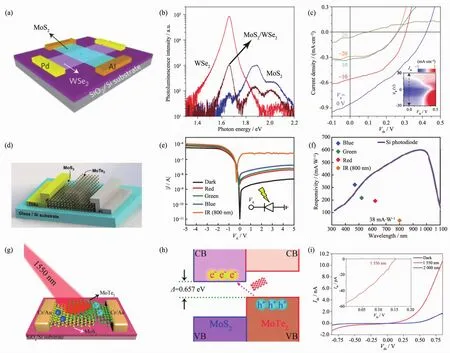
图6 (a~c)n-MoS2/p-WSe2光探测器[62];(d~f)MoS2/MoTe2光探测器[63];(g~i)MoS2/MoTe2红外光探测器[64]Fig.6 (a~c)n-MoS2/p-WSe2photodetector[62];(d~f)MoS2/MoTe2photodetector[63];(g~i)MoS2/MoTe2photodetector[64]
除了传统的剥离转移方法可以制备得到异质结构以外,原位外延生长、掺杂等方法都被用于异质结的 可 控 制 备[67-72]。
有机小分子和半导体聚合物由于结构具有更高的可调控性受到了科学家们的广泛关注。有机小分子和半导体聚合物的表面原子可以与二维材料之间形成良好的接触,从而形成类似无机范德华异质结的结构。
Liu 等[73]首 次 报 道 了 二 硫 化 钼/红 荧 烯 (MoS2/Rubrene)的有机-无机异质结构光探测器,如图8(a~c)所示。该器件制备工艺简单,将Rubrene纳米片轻置于MoS2和制备好的电极之间即可。在栅压调控下,其整流比在102~105之间可调。在较高的光功率密度下 (20 mW·cm-2), 具有 510 mA·W-1的光响应度,并且其响应速度较普通的MoS2器件快很多(约为 5 ms)。
具有高光电转换效率的有机-无机杂化钙钛矿也被应用于光探测器件上。Cheng等[74]报道了,WSe2/CH3NH3PbI3异质结,如图 8(d~f)。 为了降低钙钛矿的离子运动程度,相关测试在77 K条件下进行。尽管如此,具有异质结构的WSe2/CH3NH3PI3仍具有很高的光响应度(642 A·W-1),相较于单独的 CH3NH3PbI3(61.6 A·W-1)提高了~10倍。 与此同时具有氮化硼保护层的器件具有很快地响应速率(~500 μs)和超过7个月的使用寿命。
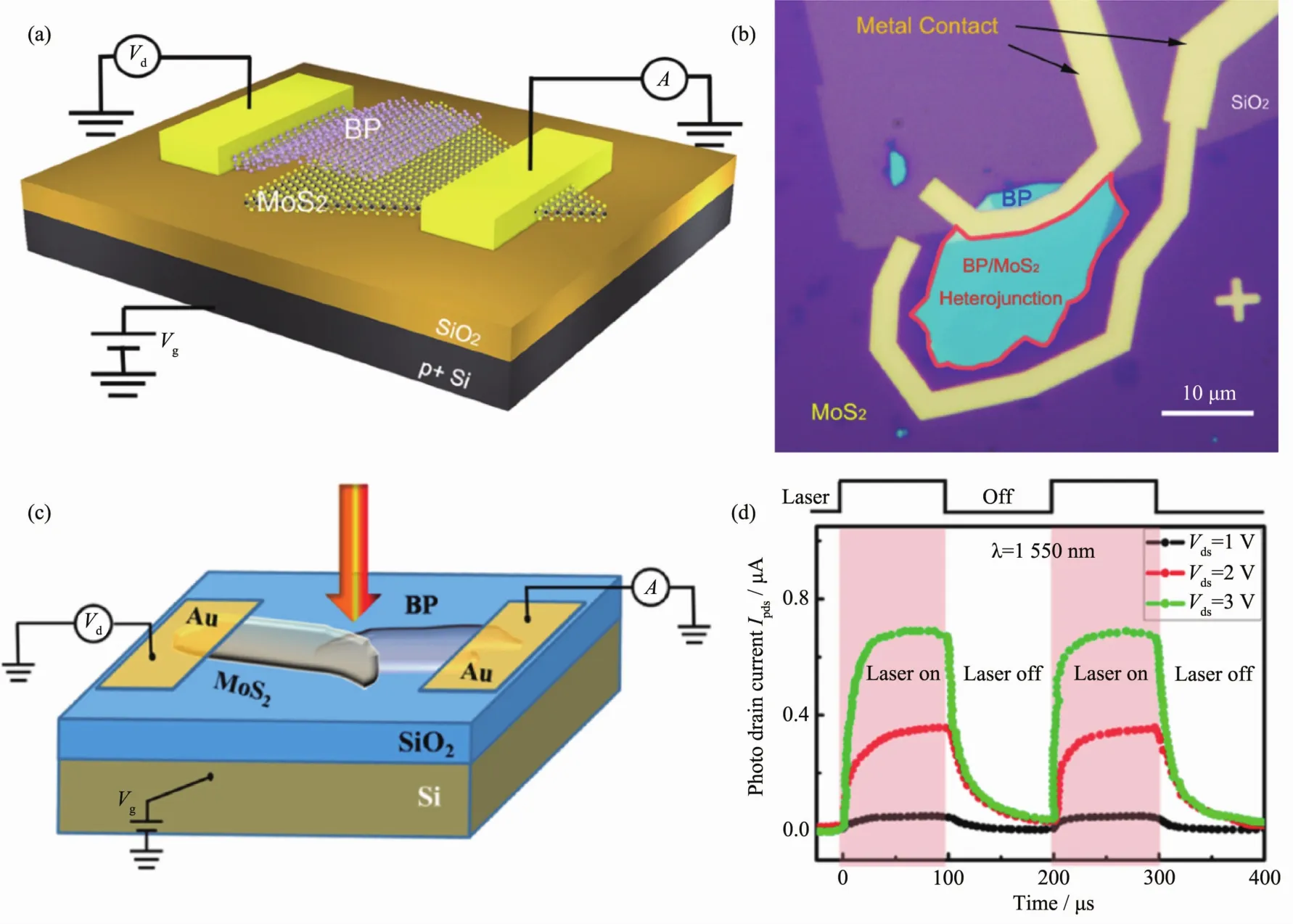
图7 (a,b)BP/MoS2 光探测器示意图[65];(c,d)BP/MoS2 在红外光下的响应[66]Fig.7 (a,b)BP/MoS2photodetector[65];(c,d)BP/MoS2photodetector′s response under 1 550 nm light[66]
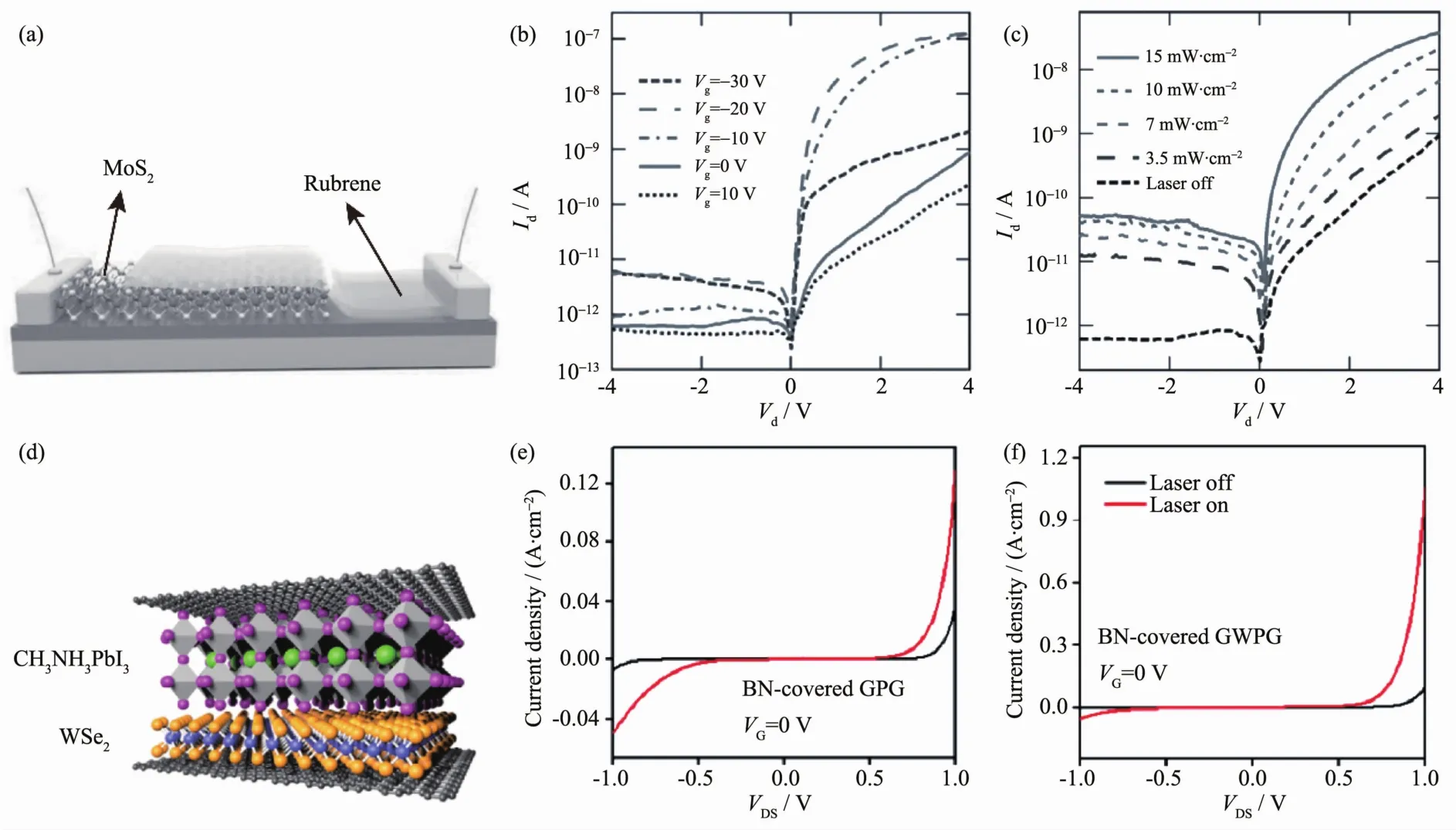
图8(a~c)MoS2/Rubrene 光探测器[73];(d~f)WSe2/CH3NH3PI3 光探测器[74]Fig.8 (a~c)MoS2/Rubrene photodetector[73],(d~f)WSe2/CH3NH3PI3photodetecto[74]
此后一系列二维有机-无机异质结光伏器件的研究工作相继被报道,为二维光探测器的设计使用提供了新的思路[75-76]。
2.3.2 敏化增强二维光探测器
尽管二维异质结构光探测器已经取得了优异的效果,但大多数异质结构器件的光增益效果并不显著,光响应度等参数也大都停留在一个较低的水平。因此如何设计得到具有宽响应光谱范围和高响应度的器件成为了二维光探测器领域亟待解决的问题。
当前具有优异光谱性能及半导体特性的零维半导体量子点材料和二维材料复合体系受到了科学家们的广泛关注。量子点除了有很好的敏化增强效果以外,由于其简单的合成和制备方法,容易被整合到柔性大规模集成器件中,因此也成为了敏化增强光探测器的一种重要方式。
2012年 Konstantatos等[77]就首次利用硫化铅(PbS)量子点敏化石墨烯,得到了具有超高光响应的光探测器,如图9(a~c)所示。其光增益效果能达到108以上,远比本征石墨烯类材料高得多。尽管其外量子效率只有25%,但是硫化铅量子点能够有效地将电子注入到高迁移率的石墨烯中,并且其表面所带来的势阱能够延长电子寿命,因而可以得到很高的光增益。制备得到的器件光响应度可达超高的~107A·W-1,归一化探测率也有 7×109Jones。 并且器件的光谱响应范围可以根据量子点的吸收光谱进行调控,实现了从950至1 450 nm人为可调控的光响应行为。同一时间,Sun等[78]也报道了不同配体修饰的PbS量子点敏化石墨烯器件具有超高的红外区域光响应,并且在低光强的895 nm红外光照下,其光响应度仍能超过106A·W-1。这些研究为今后的量子点敏化二维光探测器打下了基础。
继石墨烯之后,将MoS2作为沟道传输层进行敏化增强的工作也被报道[79]。图 9(d~f)为 PbS 量子点敏化后的MoS2光探测器。经过敏化后的MoS2光探测器的光谱响应范围和光响应度显著提高,在980 nm处器件的光响应度仍然高达~105A·W-1。在外加电场很低的情况下(3.3 mV·μm1),器件仍然具有较高的光响应值103A·W-1,进一步验证了量子点敏化能够显著提高二维光探测器的响应度和拓宽响应光谱范围的特性。
此后一系列量子点敏化二维光探测器相继报道, 如 BP、PbSe、CdS、CdSe、CuInS、Bi2Te3、 石墨烯、HgTe、SnS、SnSe、ZnO 等量子点[80-92]。 同时,对不同材料所形成的n+-n、p-n结构对光探测器性能的增强机理也做了深入详细的分析。量子点敏化二维光探测器不断发展,蕴含了巨大的潜在应用价值。
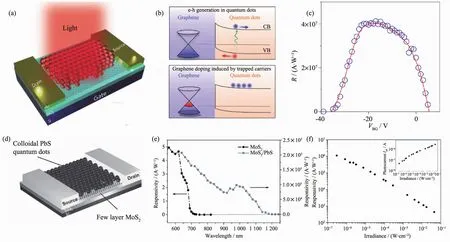
图9 (a~c)量子点敏化石墨烯光探测器[77];(d~f)量子点敏化 MoS2 光探测器[79]Fig.9 (a~c)PbS quantum dots sensitized graphene photodetector[77];(d~f)PbS quantum dots sensitized MoS2photodetector[79]
大量的实验研究表明,具有高量子效率和光谱响应范围的半导体聚合物及小分子敏化后的二维光探测器同样具有超高的光增益效果。Yu等[93]首次利用染料(罗丹明6G)对单层MoS2进行敏化,敏化后的光响应度约为敏化前的 10 倍(图 10(a,b),说明染料对MoS2的光响应性能有显著的提高。值得注意的是,染料和MoS2之间的带间跃迁能够有效地拓宽器件的光谱响应范围,截止到980 nm仍有光响应。有机分子和MoS2的这种优异的复合性能为设计合成新一代高性能敏化光探测器提供了一个平台。此后,钙钛矿、酞菁铜、红荧烯、氨丙基三乙氧基硅烷等都用来敏化二维层状材料于提高光响应度等光电性 能[93-99]。
目前在大多数有机分子敏化二维层状光探测器中,有机分子和沟道层状材料之间大多是弱的范德华作用力,因而作用不强,器件结构也不够稳定。因此科学家们开发了配位共价的敏化方法,主要包括巯基配位法[100]、重氮盐亲电配位法[101]以及金属桥配位法[102]。 Lei等[103]利用四价钛离子(Ti4+)作为 p 型试剂,可以直接和二维层状材料(MoS2、MoSe2、InSe 等)表面的孤对电子进行配位,改变其电子传输性能,进一步利用有机染料分子和Ti4+进行配位,可以在二维层状材料表面形成有效的敏化层进而提高其光响应值,如图 10(c~e)。 InSe 经 Ti4+和染料配位后,响应度是原来的~20倍。这种强的配位及共价作用为敏化剂和二维层状材料间的电子传输和界面整合提供了新的思路。
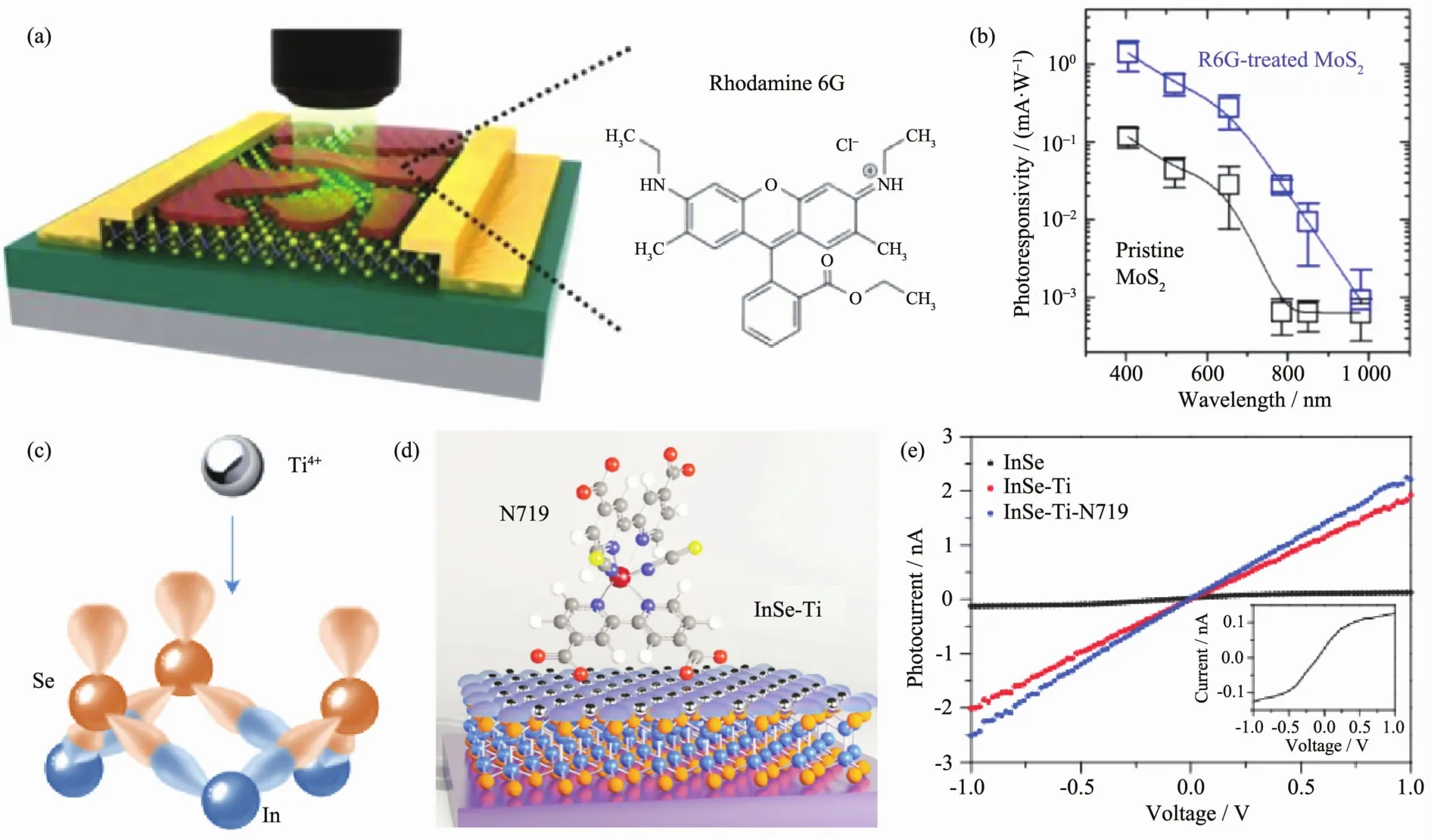
图10 (a,b)罗丹明 6G 敏化 MoS2光探测器[93];(c~e)N719、Ti4+配位敏化 InSe 光探测器[103]Fig.10 (a,b)Rhodamine6G sensitized MoS2photodetector[93];(c~e)N719,Ti4+covalent sensitized InSe photodetector[103]
2.4 二维光探测器的性能平衡
众所周知,无论是自上而下还是自下而上的方法所制备得到的二维层状材料都会存在一定的缺陷,这些缺陷在一定程度上可以捕获少数载流子,从而延长多数载流子的寿命,进而使器件具有较高的光响应度(高的光增益)。但延长载流子的寿命会导致响应时间的增加,不利于器件的快速响应。因此,二维层状材料的响应度、归一化探测率及响应速度难以同时提高,科学家们希望在其中找到一种平衡,使器件能够保持较高的工作效率。
Kufer等[104]发现,对MoS2表面进行封装之后可以有效地减少其表面的气体吸附,从而减少气体对沟道中电子的吸附,使器件性能有了很大的提高(如图 11(a~c)所示)。 并且,栅压和光照强度能够调节不同深度的势阱的占据状态(势阱越深,密度越小,寿命越长)。因此,封装的MoS2在正的栅压和低的光激发强度下,光响应度可达5×104A·W-1,但是具有相对较长的响应时间。
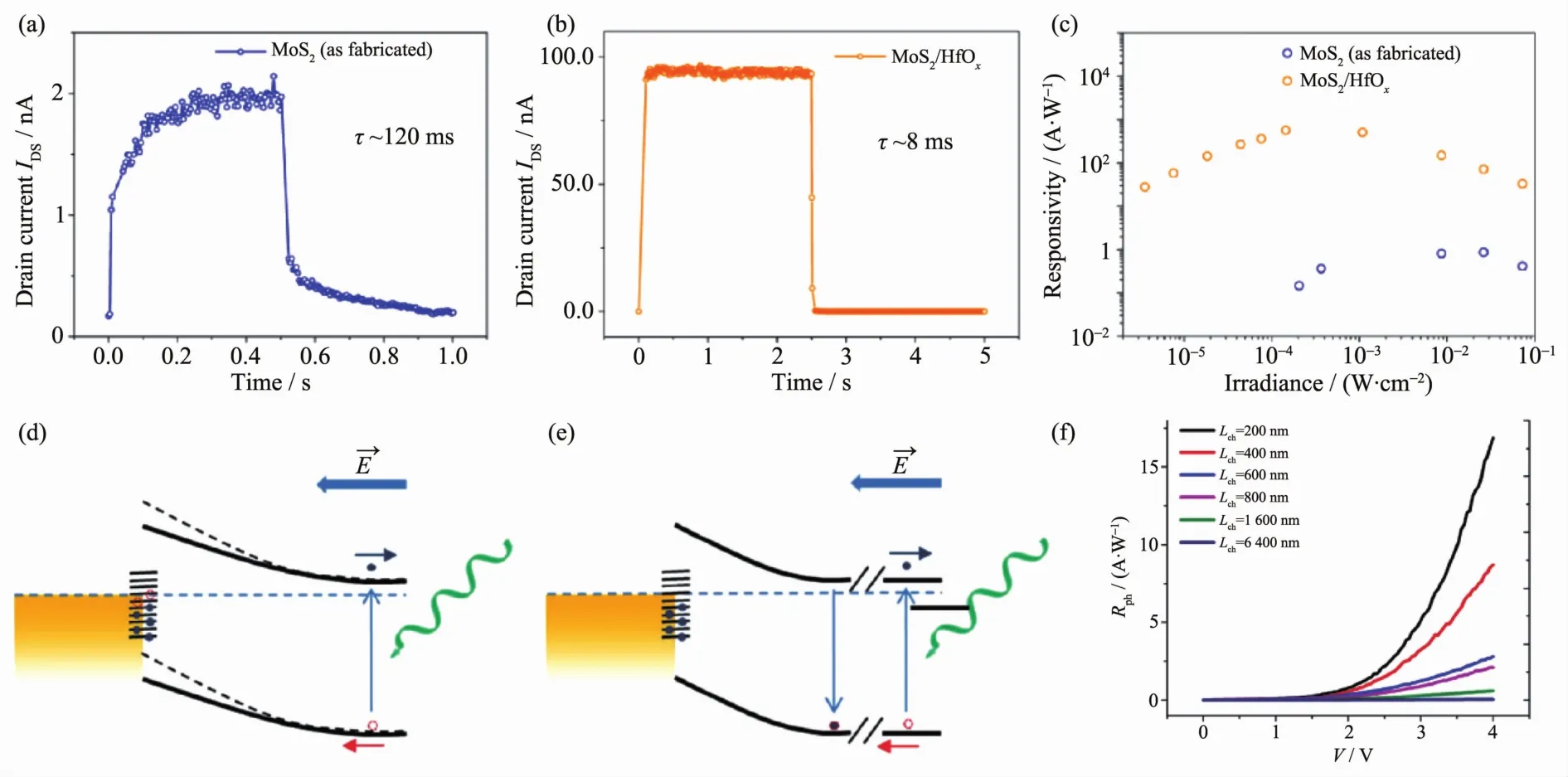
图11 (a~c)封装前后MoS2光电性能的变化[104];(d~f)沟道长度和肖特基势垒对WS2光电性能的影响[105]Fig.11 (a~c)Change of response time in MoS2photodetector before and after encapsulated[104],(d~f)Effect of gap length and schottky barrier on WS2photodetector[105]
Tsai等[106]发现如果沟道材料和电极之间能形成肖特基接触,能大大增加器件的光响应性能。在黑暗状态下,大的肖特基势垒有利于形成耗尽层和大的接触电阻,这种状态会极大地减小暗电流的大小。而在光照条件下,在界面处累积的空穴有利于势垒的降低,从而有利于形成电荷累积层,并获得大的光增益。因此,这种结构的器件可以在不改变其它参数的情况下获得很高的光增益效果,即大的光响应度、开关比和归一化探测率。且Tsai等制得的器件的响应速度是有所增加的,这可能是由大的内建电场和具有指向性的器件结构所引起的。
Fan等[105]进一步研究了沟道长度对器件响应性能的影响,他们发现短沟道的器件比长沟道的器件光响应度要提高很多(如图 11(d~f)所示)。 由于二维层状材料和电极间由肖特基结所造成的耗尽层很短,因此外加电场大多降落在该区域。然而,长沟道中的电子势阱较多,在相同光强和外加电场下,光电流和光响应度要小一个数量级以上。
为了提高本征材料的光响应性能复合结构材料得以发展,但在材料的复合敏化过程中,由于敏化剂所带来的势阱导致器件失去了原有的栅极可调特性,并且器件的响应时间会变长。因此,科学家们在敏化复合的过程中引入了界面修饰层。尽管引入的界面层会导致光电流有所下降,但因为屏蔽了表面的缺陷态,极大地降低了暗场(关闭)状态下器件的电流,使得器件的开关比大为增加。并且由于表面缺陷态的减少,器件的响应速度也得到了极大地提高。这种具有高平衡性的界面修饰层已经在PbS/MoS2及HgTe/MoS2器件中(图12)展示了其优异的调控性 能[107]。
3 二维材料家族中的新成员:石墨炔
2010年,由中国科学家首次合成并报道的新型二维半导体材料石墨炔,得到了国内外学术界的广泛关注。石墨炔材料是一类由sp和sp2杂化碳构成的本征带隙半导体,且具有较小禁带宽度、超大的平面空洞结构和极高的载流子迁移率。上述这些优异的半导体特性使石墨炔在光电转换、催化、能量存储、场发射等领域展现出了独特的优势,迅速成为新一代二维光电材料的有力竞争者[108-111]。
3.1 石墨炔的结构和特性
1987年,Baughman等[112-114]通过理论计算证明了以sp和sp2杂化所形成的石墨炔可以稳定存在,即利用若干炔键将苯环连接起来的共轭平面大分子,如图 13(a)所示。 Long 等[115]计算得到的单层石墨炔纳米片禁带宽度为0.46 eV(如图13b所示)。Smith等[116]利用密度泛函理论通过不同的计算方法预测单层的石墨炔的禁带宽度可达1.22 eV。
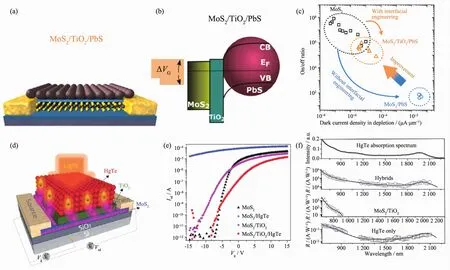
图12 (a~c)TiO2界面修饰 PbS/MoS2光探测器[107];(d~f)TiO2界面修饰 HgTe/MoS2光探测器[90]Fig.12 (a~c)PbS/TiO2/MoS2photodetector[107];(d~f)HgTe/TiO2/MoS2photodetector[90]

图13 (a)石墨炔的结构[115];(b)石墨炔的能带计算[115]Fig.13 (a)Molecule structure of graphdiyne[115];(b)Caculated band alignment of graphdiyne[115]
一系列不同的理论计算方法均预测石墨炔的禁带宽度在 0.46~1.22 eV 之间[112,117-118]。 Chen 等[119]计算了石墨炔狄拉克锥相关的电学输运性能,发现石墨炔具有极高的载流子迁移率,其空穴和电子迁移率可分别高达 4.29×105和 5.41×105cm2·V-1·s-1, 这个值甚至高于石墨烯(~3×105cm2·V-1·s-1)。 因此,理论上由于石墨炔具有合适的禁带宽度和超高的载流子迁移率,相对于传统的石墨烯、MoS2、BP等二维材料在光电子领域的应用具有更广泛的前景。
3.2 石墨炔的制备
石墨炔超高的载流子迁移率以及合适的禁带宽度使它成为了一种极具吸引力的半导体材料。自从石墨炔的概念提出来以后,国际上许多著名材料学研究小组对其可控合成的方法开展了研究。但是,早期的金属催化交叉偶联反应仅能得到小分子的石墨炔片段。因此,如何制备完整、大尺寸、高取向、高结晶度的石墨炔材料是科学界所面临的重大技术难题。
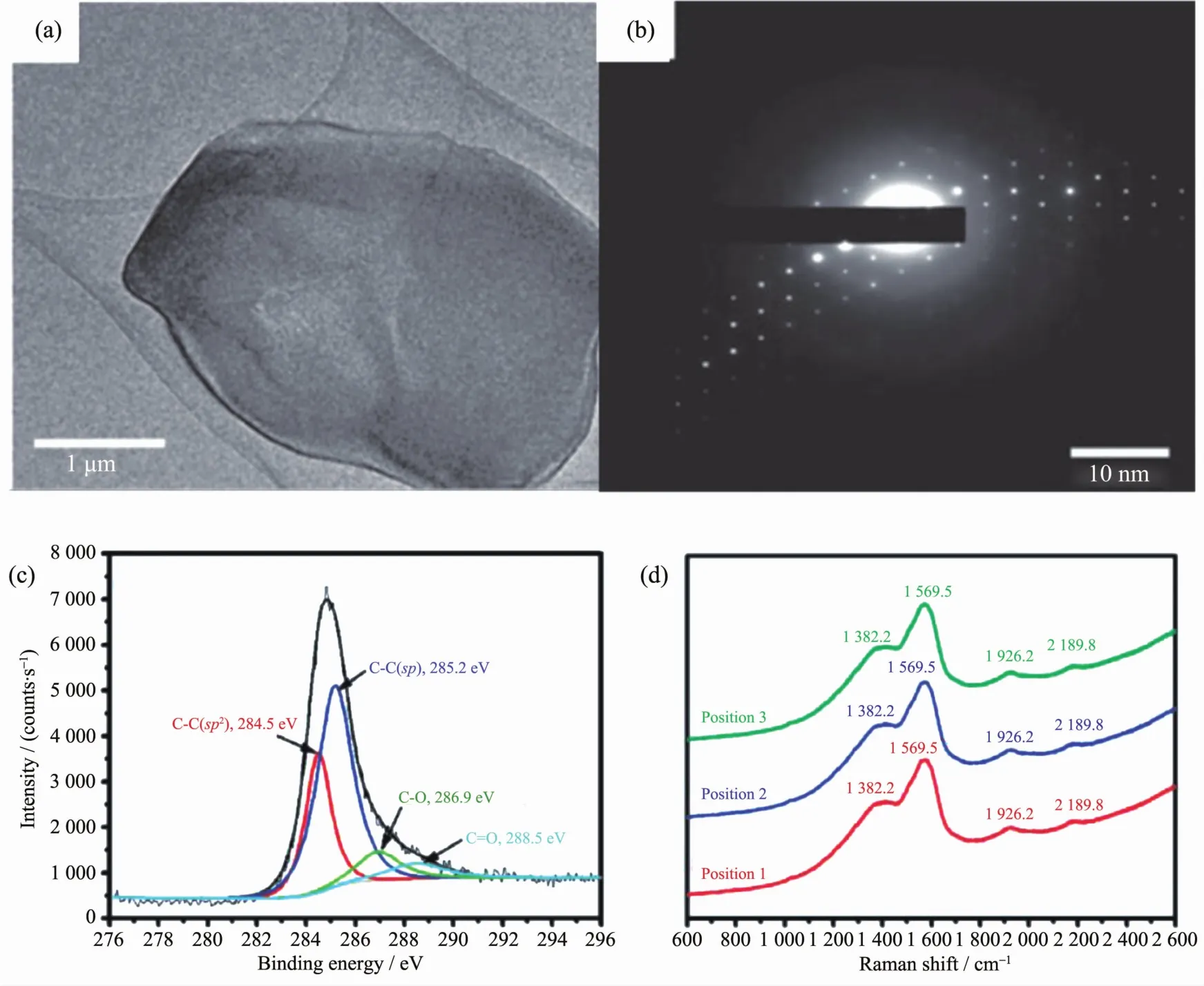
图14 (a,b)石墨炔的透射图样和电子选区衍射;(c)石墨炔的XPS图谱;(d)石墨炔的拉曼图谱[120]Fig.14 (a,b)TEM image of graphdiyne;(c)XPS spectrum of graphdiyne;(d)Raman spectrum of graphdiyne[120]
2010年中国科学院化学研究所李玉良院士课题组[120]首次提出了铜表面原位催化交叉偶联合成石墨炔的方法,成功获得了完整的单晶结构石墨炔(如图 14(a,b)所示),且其面积可达到 3.61 cm2。这是人类历史上首次制备得到大面积碳的新同素异形体石墨炔,是碳材料科学的一大进步。原位表面光电子能谱(XPS)(图 14c)确认了石墨炔的组成结构。 O1s峰的出现主要是由暴露在空气中的石墨炔吸收的少量空气造成的。而2个C1s峰285.2和284.5 eV分别为C原子的sp轨道和sp2轨道,且这2个峰的积分面积比为2∶1,这也间接说明了各苯环之间依靠2个炔键相连。石墨炔的拉曼信号峰分别为1 382.2、1 569.5、1 926.2、2 189.8 cm-1, 分别对应其 D 带、G带以及共轭二炔的伸缩振动(图14d)。这些不同的表征方式都充分证明了李玉良院士课题组成功地制备得到了高质量的石墨炔单晶材料。
Zhou等[121]采用改进型的交叉偶联方法(改变可溶性无机盐和单体的比例)让石墨炔沿着垂直方向可控生长,并且最终获得了横向尺寸为~5×103nm,厚度为15.5 nm的超薄石墨炔纳米片,如图15(a,b)所示。Matsuoka等[122]对合成方法进一步优化,在铜离子水溶液和六乙炔苯的二氯甲烷溶液界面处进行可控聚合,制备得到较大尺寸的少数层石墨炔单晶材料(>2.5×104nm),并且将厚度控制在 24 nm 以下(如图 15(c~f)所示)。 为了进一步获得层数更少的石墨炔纳米片,Matsuoka等对该方法进一步改进,如图15(g~h)所示,在醋酸铜溶液表面均匀的洒上含有六乙炔苯的苯和二氯甲烷溶液,使有机溶剂慢慢挥发,24 h后可获得横向尺寸为1~3×103nm的超薄石墨炔纳米片(厚度为3 nm),掠射小角X射线衍射表明制备得到的石墨炔纳米片具有良好的结晶性。
由于液相合成方法难以有效地控制高质量石墨炔材料的面积大小和厚度,Liu等开发了化学气相沉积法制备单层石墨炔。在液相反应中,铜被认为是一种有效的耦合催化剂,但是在表面催化耦合反应中,与铜相比银有更高的催化活性。Liu等[123]利用银箔作为基底催化生长制备得到了单层的石墨炔结构。但是通过这种方法制备的单层石墨炔纳米片是非晶结构,因此如何可控制备大面积、高质量、少层的石墨炔单晶材料仍有待进一步的研究。

图15 (a,b)石墨炔的液相合成[121];(c~f)液/液界面合成超薄石墨炔纳米片[122];(g~h)气/液界面合成超薄石墨炔纳米片[122]Fig.15 (a,b)Liquid phase fabrication of graphdiyne[121];(c~f)Liquid/Liquid interface synthesis of ultrathin graphdiyne nanosheet[122];(g~h)Gas/Liquid interface synthesis of ultrathin graphdiyne nanosheet[122]
3.3 石墨炔的应用
3.3.1 石墨炔在催化中的应用
石墨炔被认为是一种良好的催化材料,理论计算表明石墨炔在低温条件下对一氧化碳具有催化氧化功能,并且具有优秀的光、电催化能力。
Wang 等[124]利用水热法在二氧化钛(TiO2)上负载了石墨炔,负载了石墨炔的TiO2吸光能力得到了有效地改善。且在其表面,石墨炔-TiO2间的快速电荷转移也能有效地增强其光降解亚甲基蓝的能力,如图16(a~b)所示。进一步的理论计算揭示了石墨炔-TiO2之间的化学结构和电子特性。Wang等[125]利用第一性原理密度泛函理论详细计算了TiO2的不同晶面和石墨炔复合时的具体结构、电荷分离及氧化能力。 发现基于 TiO2(001),(101),(110)晶面的计算结果中,TiO2(001)-石墨炔复合材料的光催化性能较纯的 TiO2(001)及 TiO2(001)-石墨烯复合材料要优异得多。实验结果也表明TiO2(001)-石墨炔复合材料光降解亚甲基蓝的效果是纯TiO2(001)的1.67倍,是TiO2(001)-石墨烯复合材料的 1.27 倍(如图 16(c~d)所示)。此外,Li等[126]首次合成出了 β-石墨炔,与之前合成的γ-石墨炔不同的是,β-石墨炔是一种与石墨烯类似的具有零禁带宽度的材料,呈现一定的金属性。理论计算和实验表明β-石墨炔具有更高的共轭体系,因而相较于γ-石墨炔修饰的TiO2具有更好的光催化性能,如图 16(e,f)所示。 最近,Wu 等[127]利用石墨炔作为光电催化电解池中CdSe量子点和阴极之间的空穴传输层,如图17(a,b)所示。由于石墨炔良好的空穴传输能力,其在中性水溶液中的电流密度可达70 μA·cm-2,这种高的空穴传输能力有利于催化过程中CdSe上产生的空穴快速分离,并传输到阳极进行氧气的产生。因此,阴极的法拉第效率可达90%±5%。Xue等[128]采取简单的液相混合方法用氮掺杂石墨炔包裹钴纳米颗粒(CoNC/GD)。制备得到的复合CoNC/GD具有极好的稳定性,它在碱性、酸性、中性条件下分别循环36 000、38 000以及9 000次后性能几乎不衰减,远远优于Pt/C(10%)的稳定性,如图17(c,d)所示。石墨炔在催化领域展现出巨大的潜在开发价值,是一种极具应用价值的新兴催化材料。
3.3.2 石墨炔在锂电中的应用
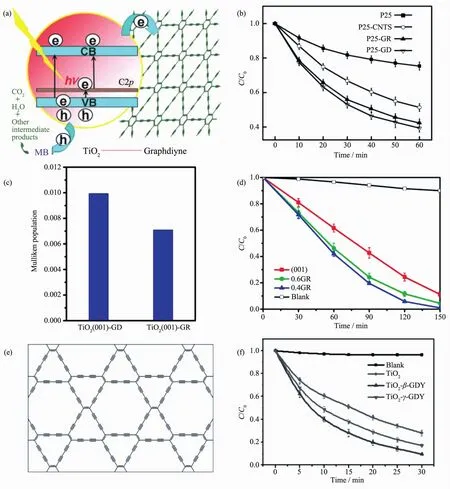
图16 (a,b)石墨炔掺杂 P25 光催化原理及性能[124];(c,d)(001)面 TiO2修饰后的电荷分布及光催化效果[125];(e,f)β-石墨炔的结构及其修饰 TiO2的光催化性能[126]Fig.16 (a,b)Mechanism and photocatalysis properties of P25 doped graphdiyne[124];(c,d)Photocatalysis proeprties and charge distribution of(001)-TiO2doped graphdiyne and graphene[125];(e,f)Structure of β-graphdiyne and its photocatalysis properties before and after TiO2doped[126]
目前研究得最广泛的是γ-石墨炔,它可以看作是在三分之一的C-C键中插入2个C≡C键,这样一来石墨炔中不但含有苯环,还兼有C≡C所构成的三角形大环(18个C原子),这就使得石墨炔的孔径大于石墨烯的孔径,让石墨炔既保有类似石墨烯的二维平面特性,同时又具备三维多孔材料的特性。此外,Sun等[129]进一步的理论计算表明,由于炔键的存在,原子密度降低,石墨炔可以以LiC3的方式吸附锂,其理论比容量的极限值可达744 mAh·g-1,是石墨烯的2倍。并且由于石墨炔大孔隙的特点,锂离子的平面扩散壁垒仅有0.18~0.84 eV,这让锂离子具有很快地扩散速率。
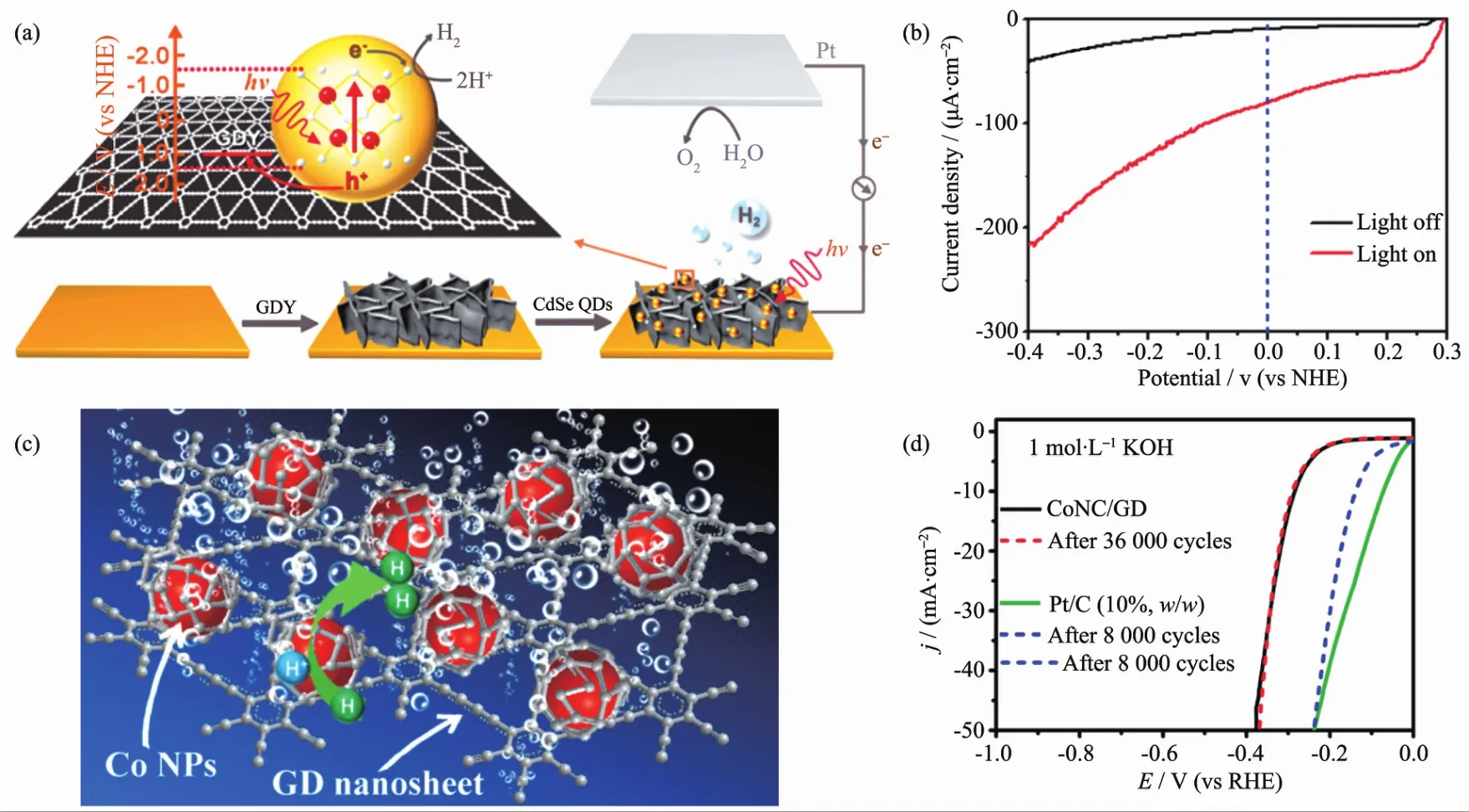
图17 (a,b)石墨炔空穴传输层CdSe量子点光电产氢[127];(c,d)CoNC/GD复合产氢及其与Pt/C循环稳定性对比[128]Fig.17 (a,b)Graphdiyne as hole transfer layer for CdSe QDs photo-electro HER[127];(c,d)Graphdiyne-Co NPs hybrid system for HER and the stability comparison to Pt/C[128]

图18 (a~c)石墨炔锂电池示意图及电容量测试曲线[130];(d~f)氮掺杂石墨炔示意图及电容量测试曲线[131]Fig.18 (a~c)Scheme of Graphdiyne as lithium battery electrode and it cycle performance[130];(d~f)Scheme of N doped Graphdiyne as lithium battery electrode and it cycle performance[131]
中国科学院化学研究所李玉良院士课题组首次实现了石墨炔的高性能储锂[130]。为了获得石墨炔储锂性能最优的条件,他们设计了3种厚度分别为10.9、22.1、30.9 μm的石墨炔锂电池。实验结果表明随着石墨炔厚度的减小,其储锂性能是增加的。在开始200圈的循环中3种不同厚度的电池容量分别可达 495、260、127 mAh·g-1。其中 10.9 μm 的电池容量已经接近石墨炔的理论极限值(744 mAh·g-1),在随后的200圈循环中逐渐趋于稳定并最终获得520 mAh·g-1的容量,即使在较大的电流密度条件下(2 A·g-1),可逆循环1 000圈后其仍能达到420 mAh·g-1的容量(如图 18(c)所示)。 这说明石墨炔不仅具有优良的储锂能力,还能快速的进行充放电。随后Zhang等[131]对石墨炔进行氮掺杂的改性,提高石墨炔对锂离子的结合能力,如图18(d~f)所示。 掺杂后的石墨炔在电流密度为200 mA·g-1条件下循环200圈后,其容量可达超高的785 mAh·g-1,实验结果甚至超过了本征石墨炔的极限容量 (744 mAh·g-1),在电流密度为400 mA·g-1条件下循环400圈后,其电池容量仍可达761 mAh·g-1,说明掺杂后的石墨炔表现出了超高的储锂性能。并且在大电流密度的实验中(1,2,4,10 A·g-1),其可逆容量仍然高达 490,443,384,299 mAh·g-1,说明氮掺杂石墨炔不但具有高的储锂稳定性还能快速的进行充放电。以上的实验结果表明石墨炔是一种非常有潜力的储锂材料。
3.3.3 石墨炔在光电器件中的应用
在光电转换器件中,器件结构设计直接影响其性能的好坏[132]。石墨炔由于其优异的传输性能,非常适合用作光电转换器件中的传输层甚至是活性层。
Xiao等[133]首次利用石墨炔对钙钛矿太阳能电池中的聚3-己基噻吩(P3HT)空穴传输层进行掺杂。拉曼光谱表明石墨炔和P3HT之间有强的π-π相互作用,而这种强的共轭作用能极大地改善电池的空穴传输性能,有效地促进活性层中空穴的提取;另一方面石墨炔有一定的散射特性,可以有效地增加器件在长波段处的吸收。因此,石墨炔掺杂后的太阳能电池效率可以达到14.58%,其平均能量转换效率较掺杂之前提高了20%(如图19(a,b)所示)。并且由于石墨炔具有极好的稳定性,器件在放置4个月之后效率仍能达到原来的90%以上。
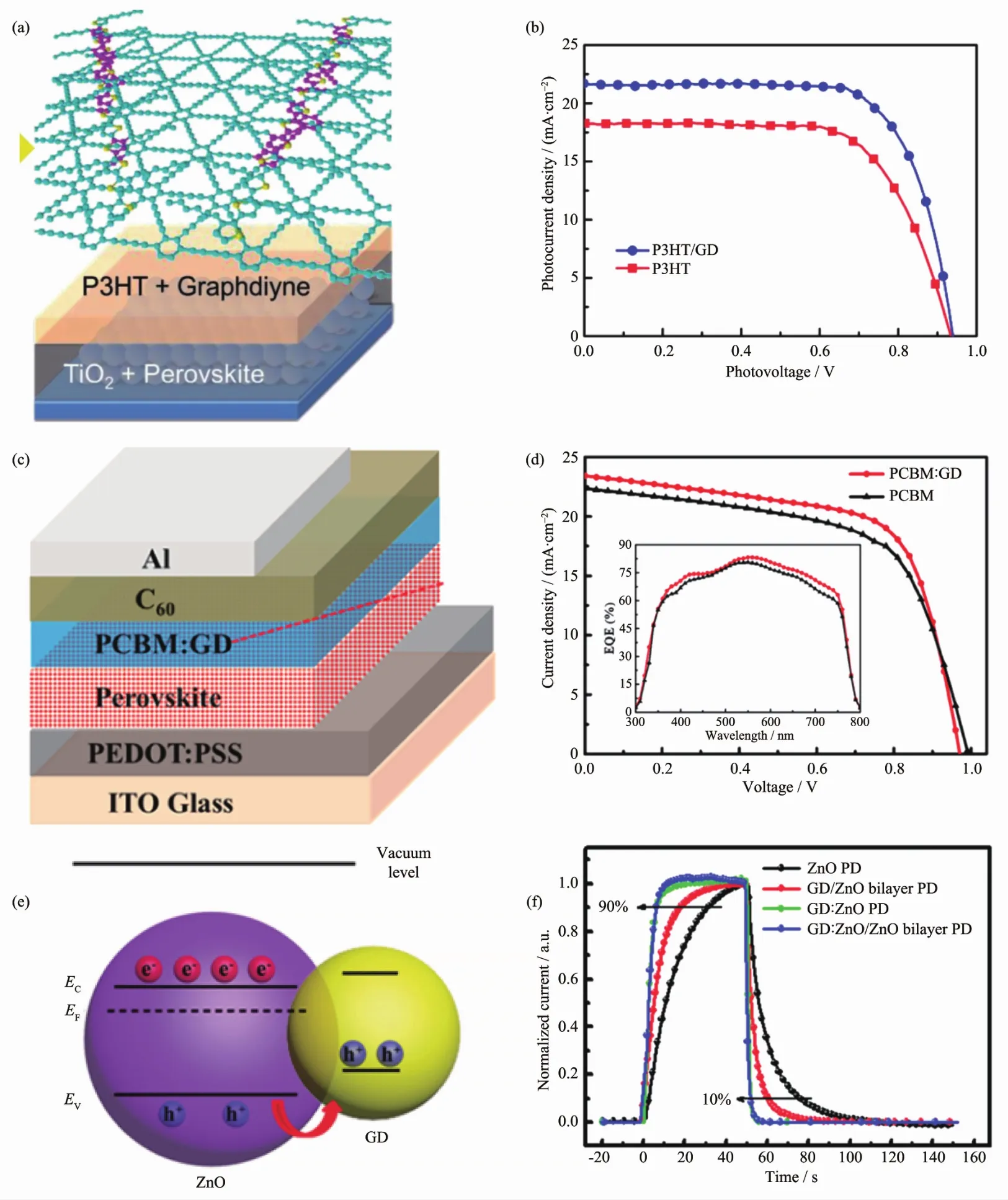
图19 (a,b)石墨炔掺杂 P3HT 太阳能电池[133];(c,d)石墨炔掺杂 PCBM 太阳能电池[134];(e,f)石墨炔掺杂 ZnO 紫外光探测器[135]Fig.19 (a,b)Graphdiyne doped P3HT solar cell[133];(c,d)Graphdiyne doped PCBM solar cell[134];(e,f)Graphdiyne doped ZnO UV photodetector[135]
类似地,Kuang等[134]用石墨炔对太阳能电池中的电子传输层[6,6]-苯基-C61-丁酸甲酯(PCBM)进行掺杂,以提高钙钛矿太阳能电池的能量转换效率,如图19(c,d)所示,目前其能量转换效率最高可达14.8%,平均能量转换效率较掺杂之前提高了28.7%。石墨炔掺杂后可以有效地覆盖光敏层中的缺陷态,提高了光敏层和电极之间的导电率,并且石墨炔还能有效的从活性层中提取电子。这一系列的因素使得太阳能电池的能量转换效率较掺杂之前有显著提高。
Jin 等[135]利用石墨炔纳米颗粒和氧化锌(ZnO)纳米颗粒复合制得了高响应的紫外光探测器,如图19(e,f)所示。在器件组装的过程中由于石墨炔纳米粒子分布于ZnO纳米粒子之间,在减少ZnO颗粒的团聚的同时也减小了ZnO纳米粒子的尺寸。但是研究表明尺寸更大的ZnO颗粒具有更高的迁移率(ZnO,1.2×10-3cm2·V-1·s-1;GD:ZnO,2.4×10-4cm2·V-1·s-1)。Jin等为了在保证器件的开关比(同时兼具小的暗电流和大的光电流),设计了石墨炔:氧化锌/氧化锌(GD∶ZnO/ZnO)双层光探测器。石墨炔对ZnO的掺杂可以有效地抑制暗电流,在光照条件下石墨炔可以捕获空穴延长电子的寿命有效地增大光电流。并且,在两层之间形成的耗尽层宽度在有无光照的条件下可以很快地调节,因此器件具有很快地响应速率。最终获得的最优条件下的器件光响应度可达1 260 A·W-1,光响应速率可达6.1/2.1 s,明显优于传统的紫外光探测器(174 A·W-1,32.1/28.7 s)。
4 结论与展望
自2011年MoS2首次被用于光探测器以来二维半导体材料在光探测器领域得到了长足的发展,目前基于MoS2的光探测器的光响应度最高可达880 A·W-1。此外,一系列二维材料(从宽带隙的GaS、GaSe到窄带隙的BP)的本征光探测器也已经得到应用。由于材料本身的限制,本征光探测器难以同时具有宽的响应光谱范围和高的响应度及快速响应速率,因此科学家们发展了敏化等方法对二维材料进行调控,使得其光谱响应范围能有所拓宽,光响应度和响应速率也有不同程度的提高。
相较于传统二维半导体材料材料,新兴的石墨炔因其理论上具有比其它传统二维材料更高的迁移率和更窄的禁带宽度而倍受关注。目前,科学家们已经成功合成出了石墨炔单晶材料,并且利用拉曼、XRD、XPS等一系列的表征方法对其结构进行了详细的研究。此外,一系列的理论计算研究也预测了石墨炔的半导体特性及其潜在的应用领域。自首次合成得到石墨炔以来,石墨炔已经实际应用到了电子、微电子、半导体、膜分离、催化、能源储存等领域,与理论计算结果相呼应。石墨炔优异的电学、化学、机械、磁性都可以通过其形貌和官能团进行相应的调控,这些优异的性能保证了石墨炔在未来二维材料研究中具有巨大的潜力。
然而目前仍然缺少能够批量制备大面积、高质量、层数可控石墨炔单晶的方法,并且石墨炔家族中,目前也只有γ-石墨二炔,β-石墨炔以及非晶的石墨炔类似物被合成出来。因此石墨炔的性质及其在光电转换等领域的潜在应用也还处于起步阶段,该领域仍有许多重要的科学问题亟待解决,在面对巨大的挑战同时也包含了巨大的机遇,相信石墨炔在未来能够成为新一代光电材料。
致谢:感谢中组部“青年千人计划”,南开大学“百名青年学科带头人”,南开大学先进能源材料化学教育部重点实验室开放基金对本项目的支持。
[1]Kroto H W,Heath J R,O′brien S C,et al.Nature,1985,318(6042):162-163
[2]Iijima S.Nature,1991,354(6348):56
[3]Mermin N D,Wagner H.Phys.Rev.Lett.,1966,17(22):1133
[4]Boehm H P.Angew.Chem.Int.Ed.,2010,49(49):9332-9335
[5]Novoselov K S,Geim A K,Morozov S V,et al.Science,2004,306(5696):666-669
[6]De Abajo F J G.Science,2013,339(6122):917-918
[7]Freitag M.Nat.Nanotechnol.,2008,3(8):455-457
[8]Novoselov K S,Jiang D,Schedin F,et al.PNAS,2005,102(30):10451-10453
[9]Ding Y,Wang Y L,Ni J,et al.Physica B,2011,406(11):2254-2260
[10]Wang Q H,Kalantar-Zadeh K,Kis A,et al.Nat.Nanotechnol.,2012,7(11):699-712
[11]Kuc A,Zibouche N,Heine T.Phys.Rev.B:Condens.Matter,2011,83(24):245213
[12]Huang Y X,Guo J H,Kang Y J,et al.Nanoscale,2015,7(46):19358-19376
[13]Jariwala D,Sangwan V K,Lauhon L J,et al.ACS Nano,2014,8(2):1102-1120
[14]Pospischil A,Mueller T.Appl.Sci.,2016,6(3):78
[15]Dhanabalan S C,Ponraj J S,Zhang H,et al.Nanoscale,2016,8(12):6410-6434
[16]Zhang H.ACS Nano,2015,9(10):9451-9469
[17]Geim A K,Novoselov K S.Nat.Mater.,2007,6(3):183-191
[18]Neto A C,Guinea F,Peres N M,et al.Rev.Mod.Phys.,2009,81(1):109
[19]Mak K F,He K,Shan J,et al.Nat.Nanotechnol.,2012,7(8):494-498
[20]Koppens F H,Mueller T,Avouris P,et al.Nat.Nanotechnol.,2014,9(10):780-793
[21]Wang H X,Wang Q,Zhou K G,et al.Small,2013,9(8):1266-1283
[22]Sun Z H,Chang H X.ACS Nano,2014,8(5):4133-4156
[23]Chhowalla M,Shin H S,Eda G,et al.Nat.Chem.,2013,5(4):263-275
[24]Eigler S,Hirsch A.Angew.Chem.Int.Ed.,2014,53 (30):7720-7738
[25]Coleman J N,Lotya M,O′neill A,et al.Science,2011,331(6017):568-571
[26]Cunningham G,Lotya M,Cucinotta C S,et al.ACS Nano,2012,6(4):3468-3480
[27]O′neill A,Khan U,Coleman J N.Chem.Mater.,2012,24(12):2414-2421
[28]Zhou K G,Mao N N,Wang H X,et al.Angew.Chem.Int.Ed.,2011,123(46):11031-11034
[29]Hanlon D,Backes C,Higgins T M,et al.Chem.Mater.,2014,26(4):1751-1763
[30]Eda G,Yamaguchi H,Voiry D,et al.Nano Lett.,2011,11(12):5111-5116
[31]Zeng Z Y,Yin Z Y,Huang X,et al.Angew.Chem.Int.Ed.,2011,50(47):11093-11097
[32]You X Q,Liu N,Lee C J,et al.Mater.Lett.,2014,121:31-35
[33]Liu N,Kim P,Kim J H,et al.ACS Nano,2014,8(7):6902-6910
[34]Reina A,Jia X T,Ho J,et al.Nano Lett.,2008,9(1):30-35
[35]Li X S,Cai W W,An J,et al.Science,2009,324(5932):1312-1314
[36]Lee Y H,Zhang X Q,Zhang W J,et al.Adv.Mater.,2012,24(17):2320-2325
[37]Song L,Ci L,Lu H,et al.Nano Lett.,2010,10(8):3209-3215
[38]Ji Q Q,Zhang Y,Zhang Y F,et al.Chem.Soc.Rev.,2015,44(9):2587-2602
[39]Yoo D,Kim M,Jeong S,et al.JACS,2014,136(42):14670-14673
[40]Xia F N,Mueller T,Lin Y M,et al.Nat.Nanotechnol.,2009,4(12):839-843
[41]Mueller T,Xia F N,Avouris P.Nat.Photonics,2010,4(5):297-301
[42]Zhang Y Z,Liu T,Meng B,et al.Nat.Commun.,2013,4:1811
[43]Hu P A,Wang L F,Yoon M,et al.Nano Lett.,2013,13(4):1649-1654
[44]Hu P A,Wen Z Z,Wang L F.ACS Nano,2012,6(7):5988-5994
[45]Yin Z Y,Li H,Jiang L.ACS Nano,2011,6(1):74-80
[46]Lopez-Sanchez O,Lembke D,Kayci M,et al.Nat.Nanotechnol.,2013,8(7):497-501
[47]Choi W,Cho M Y,Konar A,et al.Adv.Mater.,2012,24(43):5832-5836
[48]Liu F C,Shimotani H,Shang H.ACS Nano,2014,8(1):752-760
[49]Hu P A,Zhang J,Yoon M N,et al.Nano Res.,2014,7(5):694-703
[50]Lei S D,Wen F F,Ge L H,et al.Nano Lett.,2015,15(5):3048-3055
[51]Tamalampudi S R,Lu Y Y,Kumar U R,et al.Nano Lett.,2014,14(5):2800-2806
[52]Lei S D,Ge L H,Najmaei S,et al.ACS Nano,2014,8(2):1263-1272
[53]Mudd G W,Svatek S A,Ren T,et al.Adv.Mater.,2013,25(40):5714-5718
[54]Feng W,Wu J B,Li X,et al.J.Mater.Chem.C,2015,3(27):7022-7028
[55]Buscema M,Groenendijk D J,Blanter S I,et al.Nano Lett.,2014,14(6):3347-3352
[56]Tran V,Soklaski R,Liang Y,et al.Phys.Rev.B:Condens.Matter,2014,89(23):235319
[57]Eswaraiah V,Zeng Q S,Long Y,et al.Small,2016,12(26):3480-3502
[58]Wang X,Jones A M,Seyler K L,et al.Nat.Nanotechnol.,2015,10(6):517-521
[59]Zhang S,Yang J,Xu R J,et al.ACS Nano,2014,8(9):9590-9596
[60]Wu J,Koon G K W,Xiang D,et al.ACS Nano,2015,9(8):8070-8077
[61]Choi K,Lee Y T,Im S.Nano Today,2016,11(5):626-643
[62]Lee C H,Lee G H,Van Der Zande A M,et al.Nat.Nanotechnol.,2014,9(9):676-681
[63]Pezeshki A,Shokouh S H,Nazari T,et al.Adv.Mater.,2016,28(16):3216-3222
[64]Zhang K,Zhang T N,Cheng G H,et al.ACS Nano,2016,10(3):3852-3858
[65]Deng Y X,Luo Z,Conrad N J,et al.ACS Nano,2014,8(8):8292-8299
[66]Ye L,Li H,Chen Z F,et al.ACS Photonics,2016,3(4):692-699
[67]Yang S X,Wang C,Ataca C,et al.ACS Appl.Mater.Interfaces,2016,8(4):2533-2539
[68]Choi M S,Qu D,Lee D,et al.ACS Nano,2014,8(9):9332-9340
[69]Chen J Y,Zhou W,Tang W,et al.Chem.Mater.,2016,28(20):7194-7197
[70]Duan X D,Wang C,Shaw J C,et al.Nat.Nanotechnol.,2014,9(12):1024-1030
[71]Li B,Huang L,Zhong M Z,et al.Adv.Electron.Mater.,2016,2(11):1600298
[72]Xue Y Z,Zhang Y P,Liu Y,et al.ACS Nano,2016,10(1):573-580
[73]Liu F C,Chow W L,He X X,et al.Adv.Funct.Mater.,2015,25(36):5865-5871
[74]Cheng H C,Wang G,Li D,et al.Nano Lett.,2016,16(1):367-373
[75]Jariwala D,Howell S L,Chen K S,et al.Nano Lett.,2016,16(1):497-503
[76]Velez S,Ciudad D,Island J,et al.Nanoscale,2015,7(37):15442-15449
[77]Konstantatos G,Badioli M,Gaudreau L,et al.Nat.Nanotechnol.,2012,7(6):363-368
[78]Sun Z H,Liu Z K,Li J H,et al.Adv.Mater.,2012,24(43):5878-5883
[79]Kufer D,Nikitskiy I,Lasanta T,et al.Adv.Mater.,2015,27(1):176-180
[80]Qiao H,Yuan J,Xu Z Q,et al.ACS Nano,2015,9(2):1886-1894
[81]Schornbaum J,Winter B,Schiessl S P,et al.Adv.Funct.Mater.,2014,24(37):5798-5806
[82]Chen C Y,Qiao H,Lin S H,et al.Sci.Rep.,2015,5:11830
[83]Huang Y,Zhan X Y,Xu K,et al.Appl.Phys.Lett.,2016,108(1):013101
[84]Jia Z Y,Xiang J Y,Wen F S,et al.ACS Appl.Mater.Interfaces,2016,8(7):4781-4788
[85]Ra H S,Kwak D H,Lee J S.Nanoscale,2016,8(39):17223-17230
[86]Wang W,Niu X Y,Qian H L,et al.Nanotechnology,2016,27(50):505204
[87]Zheng W,Feng W,Zhang X,et al.Adv.Funct.Mater.,2016,26(16):2648-2654
[88]Ahmad R,Srivastava R,Yadav S,et al.J.Phys.Chem.Lett.,2017,8(8):1729-1738
[89]Hu C,Dong D D,Yang X K,et al.Adv.Funct.Mater.,2017,27(2):1603605
[90]Huo N J,Gupta S,Konstantatos G.Adv.Mater.,2017,29(17):1606576
[91]Jia Z,Li S,Xiang J,et al.Nanoscale,2017,9(5):1916-1924
[92]Nazir G,Khan M F,Akhtar I,et al.RSC Adv.,2017(27):16890-16900
[93]Yu S H,Lee Y B,Jang S K,et al.ACS Nano,2014,8(8):8285-8291
[94]Pak J,Jang J,Cho K,et al.Nanoscale,2015,7(44):18780-18788
[95]Cho E H,Song W G,Park C J,et al.Nano Res.,2014,8(3):790-800
[96]Kang D H,Kim M S,Shim J,et al.Adv.Funct.Mater.,2015,25(27):4219-4227
[97]Lee Y,Kwon J,Hwang E,et al.Adv.Mater.,2015,27(1):41-46
[98]Kang D H,Pae S R,Shim J,et al.Adv.Mater.,2016,28(35):7799-7806
[99]Ma C,Shi Y M,Hu W J,et al.Adv.Mater.,2016,28(19):3683-3689
[100]Chou S S,De M,Kim J,et al.JACS,2013,135(12):4584-4587
[101]Knirsch K C,Berner N C,Nerl H C,et al.ACS Nano,2015,9(6):6018-6030
[102]Backes C,Berner N C,Chen X,et al.Angew.Chem.Int.Ed.,2015,54(9):2638-2642
[103]Lei S D,Wang X F,Li B,et al.Nat.Nanotechnol.,2016,11(5):465-471
[104]Kufer D,Konstantatos G.Nano Lett.,2015,15 (11):7307-7313
[105]Fan Y,Zhou Y Q,Wang X C,et al.Adv.Opt.Mater.,2016,4(10):1573-1581
[106]Tsai D S,Liu K K,Lien D H,et al.ACS Nano,2013,7(5):3905-3911
[107]Kufer D,Lasanta T,Bernechea M,et al.ACS Photonics,2016,3(7):1324-1330
[108]HUANG Chang-Shui(黄长水),LI Yu-Liang(李玉良).Acta Phys.-Chim.Sin.(物理化学学报),2016,32(6):1314-1329
[109]LI Yong-Jun(李勇军),LI Yu-Liang(李玉 良).Acta Polym.Sin.(高分子学报),2015,2:147-165
[110]CHEN Yan-Huan(陈彦焕),LIU Hui-Biao(刘辉彪),LI Yu-Liang(李玉良).Chin.Sci.Bull.(科学通报),2016,61(26):2901
[111]QIAN Xue-Min(钱学旻),LIU Hui-Biao(刘辉彪),LI Yu-Liang(李玉良).Chinese J.Inorg.Chem.(无机化学学报),2014,30(1):62-74
[112]Baughman R,Eckhardt H,Kertesz M.J.Chem.Phys.,1987,87(11):6687-6699
[113]Baughman R,Zakhidov A A,De Heer W A.Science,2002,297(5582):787-792
[114]Coluci V,Galvao D,Baughman R.J.Chem.Phys.,2004,121(7):3228-3237
[115]Long M Q,Tang L,Wang D,et al.ACS Nano,2011,5 (4):2593-2600
[116]Van Miert G,Juricˇic'V,Smith C M.Phys.Rev.B:Condens.Matter,2014,90(19):195414
[117]Narita N,Nagai S,Suzuki S,et al.Phys.Rev.B:Condens.Matter,1998,58(16):11009
[118]Luo G F,Qian X M,Liu H,et al.Phys.Rev.B:Condens.Matter,2011,84(7):075439
[119]Chen J M,Xi J Y,Wang D,et al.J.Phys.Chem.Lett.,2013,4(9):1443-1448
[120]Li G X,Li Y L,Liu H B,et al.Chem.Commun.,2010,46(19):3256-3258
[121]Zhou J Y,Gao X,Liu R,et al.J.Am.Chem.Soc.,2015,137(24):7596-7599
[122]Matsuoka R,Sakamoto R,Hoshiko K,et al.J.Am.Chem.Soc.,2017,139(8):3145-3152
[123]Liu R,Gao X,Zhou J,et al.Adv.Mater.,2017,29 (18):1604665
[124]Wang S,Yi L X,Halpert J E,et al.Small,2012,8(2):265-271
[125]Yang N L,Liu Y Y,Wen H,et al.ACS Nano,2013,7(2):1504-1512
[126]Li J Q,Xie Z Q,Xiong Y,et al.Adv.Mater.,2017,29(19):1700421
[127]Li J,Gao X,Liu B,et al.J.Am.Chem.Soc.,2016,138(12):3954-3957
[128]Xue Y R,Li J F,Xue Z,et al.ACS Appl.Mater.Interfaces,2016,8(45):31083-31091
[129]Sun C,Searles D J.J.Phys.Chem.C,2012,116(50):26222-26226
[130]Huang C S,Zhang S L,Liu H B,et al.Nano Energy,2015,11:481-489
[131]Zhang S L,Du H P,He J J,et al.ACS Appl.Mater.Interfaces,2016,8(13):8467-8473
[132]GUO Wen-Ming(郭文明),ZHONG Min(钟敏).Chinese J.Inorg.Chem.(无机化学学报),2017,33(7):1097-1118
[133]Xiao J Y,Shi J J,Liu H B,et al.Adv.Energy Mater.,2015,5(8):1401943
[134]Kuang C Y,Tang G,Jiu T G,et al.Nano Lett.,2015,15(4):2756-2762
[135]Jin Z W,Zhou Q,Chen Y H,et al.Adv.Mater.,2016,28(19):3697-3702
Two-Dimensional Semiconducting Materials and Devices:from Traditional Two-Dimensional Optoelectronic Materials to Graphdiyne
HUANG Yan-Min1YUAN Ming-Jian*,1LI Yu-Liang2
(1College of Chemistry,Key Laboratory of Advanced Energy Materials Chemistry(Ministry of Education),Nankai University,Tianjin 300071,China)
(2CAS Key Laboratory of Organic Solids,Beijing National Laboratory for Molecular Sciences(BNLMS),Institute of Chemistry Chinese Academy of Sciences,Beijing 100080,China)
Two-dimensional (2D)semiconducting materials have been widely used in optoelectronic devices,catalysis,biosensors and other fields due to their unique molecular structure and electronic properties,herein have attracted lots of attention.In this review,the development of traditional 2D materials as well as novel 2D material graphdiyne has been summarized.The review focused on the application of traditional 2D materials in the field of photodetector,investigated the influence of different material systems and their device structures on the performance of the photodetector.In addition,we further described the synthesis and application of graphdiyne.Finally,we summarized the opportunities and challenges of traditional 2D materials and graphdiyne in the application of energy conversion devices.
two-dimensional materials;graphdiyne;optoelectronic materials and devices
O613.71;O614.61;O616
A
1001-4861(2017)11-1914-23
10.11862/CJIC.2017.265
2017-08-02。收修改稿日期:2017-10-19。
国家自然科学基金(No.21771114)资助项目。
*通信联系人。E-mail:yuanmj@nankai.edu.cn
