含TEMPO配合物的合成、表征、谱学性质及光猝灭机理
2017-11-01陈可先赵琛烜李浩然
尹 璐 梁 程 陈可先 赵琛烜 姚 加 李浩然,,*
(1浙江大学化学系,浙大-新和成联合研发中心,杭州 310027;2浙江大学化学工程与生物工程学院,杭州 310027;3浙江工商大学食品与生物工程学院,杭州 310018)
含TEMPO配合物的合成、表征、谱学性质及光猝灭机理
尹 璐1梁 程2陈可先3赵琛烜1姚 加1李浩然1,2,*
(1浙江大学化学系,浙大-新和成联合研发中心,杭州 310027;2浙江大学化学工程与生物工程学院,杭州 310027;3浙江工商大学食品与生物工程学院,杭州 310018)
通过由2,2,6,6-四甲基哌啶-氮-氧化物(TEMPO)自由基修饰的三联吡啶配体与二价金属铂盐反应,合成得到一种新型的金属配合物,[Pt(terpy-TEMPO)Cl]Cl·H2O·CH3OH (terpy指2,2ʹ:6ʹ,2ʹ-三联吡啶)。此配合物由于TEMPO自由基的作用呈现高效率的光猝灭现象。X衍射单晶数据证实此配合物的分子结构信息。利用紫外、荧光及电子顺磁共振光谱等谱学手段探讨了该配合物的紫外吸收、发射及电子顺磁共振(EPR)光谱性质。[Pt(terpy-TEMPO)Cl]Cl·H2O·CH3OH的室温紫外吸收光谱表明,此配合物有两个典型的紫外吸收波段,强吸收段和次强吸收段,分别来源于配体到配体的跃迁(MLCT),金属到配体的跃迁(LLCT)。另外,[Pt(terpy-TEMPO)Cl]Cl·H2O·CH3OH的室温固体荧光光谱表明,TEMPO的单电子能有效地猝灭三联吡啶铂的荧光发射。我们对此猝灭机理进行了详细合理的阐述,并通过高斯09软件包对配合物的能隙和能带进行了量化计算,结果进一步证明配合物体系中的TEMPO单电子能极大的影响最高占有分子轨道(HOMO)与最低未占分子轨道(LUMO)之间的能级差,从理论上解释了三联吡啶铂配合物的光猝灭的光学性质与分子结构之间的关系。EPR结果表明,稳定自由基上接上金属配合物,不影响自由基A值和g值(A值指自由基超精细耦合常数,g值指自由基的g因子),但影响自由基转动、弛豫时间。
三联吡啶铂配合物;氮氧自由基;合成;光致发光;电子顺磁共振光谱
1 Introduction
Free radicals are important in biological1,2and environmental process3, or catalytic reaction4. 2,2,6,6-Tetramethyl-1-piperridinyloxy (TEMPO) radical, an important class of stable free radicals with one unpaired electron5, has been widely employed as a mild and selective primary catalyst. It is well known that TEMPO could catalyze the oxidation of alcohols in the presence of co-oxidants6,7. More importantly, TEMPO could be applied as a typical paramagnetic species in the quenching of photo-excited molecules. Takeuchi, Ishii and other groups demonstrated that the combination of chromophoric moieties and TEMPO could provide the direct information of spin-sublevel dependence of quenching the excited singlet state (S1) and triplet state (T1) metalloporphyrins(MPs) or metallophthalocyanines (MPcs)8,9. Recently, Blough,et al., Ishii, et al., and other groups reported the photo-induced population transfer (PIPT) in which the TEMPO was covalently coupled to a chromophore10−12.
As a kind of luminescent material, the square-planar coordination geometry of d8Pt(II) complexes possess intriguing spectroscopic, luminescence and anticancer properties13−16. Some Pt(II) terpyridine complexes have three ordered intraligand charge transfer, ligand-to-ligand charge transfer, and metal-to-ligand charge transfer states in a single mononuclear17.The promising luminescence exhibited by these complexes was attributed mainly to the dπ(Pt) → π*(N-N-N)3MLCT excited state18,19. Diversified pendants can be attached to the central metal Pt(II)20−22or modified on the ring of terpyridine23−26to construct new structures. In these cases, the nature of the auxiliary ligand and counterion of the terpyridyl chelator would quench such3MLCT excited states in the presence of a low-lying non-emissive d-d ligand field (LF) or ligand-toligand charge transfer (LLCT) states27. Furthermore, the Lewis bases can attack the open coordination sites of Pt(II) terpyridine complex in the excited state resulting in exciplex quenching28,29,and even excimer quenching would occur in the fluid solution30,31. However, the luminescence quenching of Pt(II)terpyridine complex with the TEMPO substitution has seldom been reported. It was shown in our preliminary experiments that the ascorbic acid could turn on the luminescence in quenched luminescence of Pt(II) terpyridyl- TEMPO derivative system. To the best of our knowledge, few reports related to the synthesis of the metallobiomolecular of Pt(II) complex covalently linked with TEMPO radical have been reported. So in theory, Pt(II) terpyridine tethering TEMPO complex may act as both a transition metal catalyst and bi-functional probe,which could be applied as a precursor of photoluminescence and an EPR probe. On the analysis of the conversion of EPR signal and luminescence, more information about single electron behaviour would be achieved in the process of coordination between substrates and transition metal, which could explain the TEMPO-catalyzed oxidation or cytotoxic mechanism more clearly.
Herein, we report the synthesis of a new kind of Pt(II)complex with the ligand of 2,2,6,6-tetramethyl-4-(2,2ʹ:6ʹ2ʹ-terpyridin-4ʹ-yloxy)piperidin-1-oxyl (L), in which the incorporation of TEMPO with the terpyridine derivative framework can significantly perturb the luminance and electron transition.Furthermore, the intramolecular paramagnetic quenching of singlet state by nitroxide radicals is found to be highly efficient.In this case of photoextinction effect, upon reaction with ascorbic acid to consume nitroxide radicals, the fluorescence would restore. The changes in fluorescence of the synthetic Pt(II) complex resemble to other “on/off sensor” analogous system, and can readily be applied to biological systems32.
2 Experimental
2.1 Materials and reagents
Dimethylformamide (DMF), dimethylsulfoxide (DMSO),(purity ≥ 99%, Super Dry, water ≤ 3 × 10−5(mass fraction));ether (concentration purity ≥ 99.5%); menthol (purity ≥ 99.5%),were purchased from Aladdin. Ultra-pure water was used for the preparation of all solutions. DMF was dried by distillation from sodium wire/benzophenone. Commercial K2PtCl4(Sigma Aldrich, purity ≥ 99%), 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (Sigma Aldrich, purity ≥ 98%),2,2ʹ:6ʹ,2ʹ-terpyridine (Sigma Aldrich, purity ≥ 98%), and 4ʹ-chloro-2,2ʹ:6ʹ,2ʹ-terpyridine (Sigma Aldrich, purity ≥ 98%)were used as received. All manipulations involving organometallic compounds were carried out in air atmosphere without pre-purified nitrogen or welding-grade argon using standard techniques for handling air-sensitive compounds, Except for special instructions Elemental analyses (CHN) were determined using a Vario MICRO cube instrument. X-ray crystal structure analyses were performed by using a Gemini A Ultra instrument. Structure solution and refinement was accomplished using SHELXL-97. CCDC 1063015 (compound 2) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road,Cambridge CB21EZ, UK; Fax: +44-1223-336-033; Email:de-posit@ccdc.cam.ac.uk). The PL and absorption spectra were measured using an Edinburgh instruments FLS 920 spectrometer and an analytic Jena S600 UV/Vis spectrophotometer. EPR spectrometer used was a computer controlled X-band (9.5 GHz)EPR spectrometer (Bruker A300) equipped with a variable temperature control unit (Bruker ER 4131VT Variable Temperature Accessory, which can command the temperature with an accuracy of ±0.1 K at the site of the sample). Typical ESR parameters were as follows: 3508 G center field; 60 G sweep width; 9.439 GHz microwave frequency; 15.99 mW power; 1.59 × 103receiver gain; modulation frequency of 100 kHz; modulation amplitude of 1 G; with the conversion time being 42 msec and time constant being 10.24 msec with 1 X-scans for each 6144 point spectrum.
Dichloro (l,5-cyclooctadiene) Pt(II)[Pt(COD)Cl2] was synthesized following the literature33. Yield 99%, Anal. Calc.(%) for C8H12Cl2Pt: C, 25.67; H, 3.21. Found (%): C, 25.56; H,3.19. The dichlo-ro(l,5-cyclooctadiene) Pt(II) was used without recrystallization in the syntheses of the following Pt(II)complexes.
[Pt(terpy)Cl]·Cl·2H2O (compound 1) (Scheme 1) was synthesized according to the method reported in the literature34.Yield 95%, ESI-MS: [M-Cl]+464.10; Anal. Calcd (%) for C15H15N3O2Cl2Pt: C, 33.62; H, 2.80; N, 7.85. Found (%): C,33.56; H, 2.81; N, 7.89.
2.2 Experimental and synthesis procedures
2.2.1 Synthesis of 2,2,6,6-tetramethyl-4-(2,2′:6′,2′-terpyridin-4′-yloxy)-piperidi-1-oxyl (L, terpy-TEMPO)
Ligand L was prepared by a reported procedure35: To a suspension of freshly ground KOH (2.64 g, 47.2 mmol) in DMSO (35 mL) was added 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (2.00 g, 11.8 mmol), followed by 4ʹ-chloro-2,2ʹ:6ʹ,2ʹ-terpyridine (3.33 g, 11.8 mmol). The mixture was stirred at 50 °C for 22 h, and then quenched with an equal volume of water to afford a pink solid which was dried over under reduced pressure at 50 °C. Recrystallization from hot hexanes gave feathery pale pink needles. Yield 3.9 g, 81%. M.P.128−130 °C. ESI-MS: [M+H]+404.01, 426.13. Anal. Calcd (%)for C12H27N4O2: C, 71.44; H, 6.74; N, 13.90. Found (%): C,71.09; H, 6.72; N, 13.87.
2.2.2 Synthesis of
[Pt(terpy-TEMPO)Cl]·Cl·H2O·CH3OH
(compound 2)
Method 1: To a suspension of [Pt(COD)Cl2] (1.50 g, 4.00 mmol) in methanol (100 mL), Ligand L (1.61 g, 4.00 mmol)dissolved in 20 mL methanol was added with stirring and the mixture warmed at 50 °C on oil bath. After 15 min all the pale yellow suspension liquid turned to bright yellow, and stirred for 20 h until completion of the reaction, then was cooled at room temperature. Equal volume of ethyl ether was added, in this case, the product was precipitated from the solution, and then filtered to get filter residue, washed with methanol, diethyl ether three times (3 × 50 mL), solvent was then removed under reduced pressure leaving a bright yellow tiny needle, which was collected and vacuum oven dried overnight. Yield: 2.68 g,95%. ESI-MS: [M-Cl]+634.5. Anal. Calc. (%) for C25H33Cl2N4O5Pt: C, 41.72; H, 4.62; N, 7.79. Found (%): C,41.63; H, 4.64; N, 7.70. Method 2: to a suspension of[Pt(DMSO)2Cl2] (1.50 g, 4.00 mmol) in methanol (100 mL),Ligand L (1.68 g, 4.00 mmol) dissolved in 20 mL methanol was added (cis-[Pt(DMSO)2Cl2] was synthesized according to relevant reference36). In this case, however, except for the desired compound 2, the salt [Pt(terpy-TEMPO)Cl][Pt(DMSO)Cl3] (compound 4) was also formed. The compound 4 seems to be total if the reaction time was increased to 20 h. The failure of obtaining the only [Pt(terpy-TEMPO)Cl]+is a consequence of the almost complete insolubility of 4, which subtracts the [Pt(DMSO)Cl3]−anion formed by reaction (1). Therefore, method 2 is not as good as method 1, we employed method 1 at last.Cis-[Pt(DMSO)2Cl2] + Cl−→ [Pt(DMSO)Cl3]−+ DMSO (1)
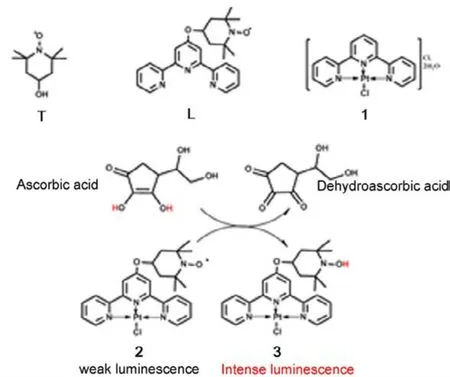
Scheme 1 Molecular structures of the 2,2,6,6-tetramethyl-1-piperridinyloxy (TEMPO) radical (T),complex ligand L, [Pt(terpy)Cl]+ derivatives, and the proposed reaction mechanism of compound 2 with ascorbic acid.
2.2.3 Synthesis of [Pt(terpy-TEMPOH)Cl]·Cl·H2O·CH3OH(compound 3)
Compound 3 was synthesized by means of “one pot process”,and all reactions were performed under an inert atmosphere of nitrogen. The ascorbic acid (0.21 g, 1.2 mmol) was added to a stirred solution of terpy-TEMPO (L, 0.16 g, 0.4 mmol) in 20 mL methanol. The resultant solution was stirred at room temperature for 3 h. Pt(COD)Cl2(0.15 g, 0.4 mmol) dissolved in methanol was dropwise added to the reaction mixture via syringe, which turned out light yellow precipitation slowly and was then stirred for another 2 h in 50 °C. The product was isolated, washed with deionized water (3 × 20 mL), methanol(3 × 20 mL), and dried in vacuum. Yield 80%. ESI-MS:[M-Cl]+635.2. Anal. Calc. for C25H34Cl2N4O5Pt: C, 41.67; H,4.76; N, 7.78. Found: C, 41.60; H, 4.69; N, 7.76.
As a final note on the complex 3, it is a kind of unstable complex, and is more inclined to transform into complex 2, so NMR data of complex 3 is difficult to obtain.
Complex 3 was stored in the anhydrous glove box under nitrogen atmosphere. The solid film layer of the complex 3 was prepared as follow: firstly, dissolved in degassed DMF solution(10−3mol·L−1); secondly, the homogeneous solution (about 0.1 mL) was dropped onto a glass sheet of 1 cm × 1 cm; finally, let it dry in flowing N2, forming a uniform sample spot, and then the glass sheet was transferred to the sealed vessel. Before we manipulated fluorescence spectrophotometry detection, the glass sheet was quickly transferred to the sample tank,subsequent purged air from the sample tank system to ensure that there is no oxygen or water prior to inject dry nitrogen. In this operation, we can guarantee that the data of fluorescence test and other experiments are reliable, repeatable.
Additionally, the solid film layers of complexes 1 and 2 were prepared in a similar method to complex 3, except that rigorous conditions (no anhydrous anaerobic) were not necessary.
3 Results and discussion
3.1 Synthesis and characterization
In this work, (terpy)-TEMPO (L) was prepared by the reaction of 4ʹ-choro-2,2ʹ:6ʹ,2ʹ-terpyridine (4-Cl-terpy) with 4-OH-TEMPO in the presence of KOH in DMSO solvent35.Chelation was carried out between K2PtCl4and 1,5-cyclooctadiene(COD) to afford the Pt(COD)Cl218. The reaction of Pt(COD)Cl2with L in methanol under reflux for 2 h gave compound 2, which was isolated as the red-orange solid in 95%yield (see Fig.S1, Supporting Information). Compound 2 was characterized by ESI-MS and elemental analysis. The ESI-MS showed that the [Pt(terpy-TEMPO)Cl]+molecular ion in CH3OH, revealed peak clusters centred at m/z 634.2 (M+, 100%)(Fig.S2, Supporting Information). We confirmed the composition of complex 2 by the X-ray crystallography, and further information was given by the TGA analysis (Fig.S3,Supporting Information). The X-ray structure provided evidence for the participation of one molecule of H2O and one molecule of CH3OH from the outer sphere in the crystallization of the compound 2 (see Fig.S4, Table S1, Supporting Information for details). It was found from the TGA result(Fig.S3, Supporting Information) that compound 2 lost its relative mass at about 200 °C due to the loss of a small amount of H2O and CH3OH, then lost sharply at 250−550 °C, and started to level off at high temperature (> 800 °C).
3.2 Crystal structure of compound 2
Crystal structure of compound 2 was obtained by layering diethyl ether into a mixed solvent of dichloromethane and methanol. In the crystal structure (Fig.1), the platinum atom adopted an approximately square planar geometry. This structure consisted of a monomeric [PtII(terpy-TEMPO)(Cl)]+cation, a chloride anion, and co-crystallized solvent molecules.The selected interatomic distances and angles were given in the Supporting Information. The metal center in the cationic complex of compound 2 adopted a very lightly distortedsquare-planar geometry with three pyridines nitrogen of terpyridine, and the chloride anion. The N―Pt―N angle was analyzed as ~80.7° which was typical in other related complexes37. The important bond distances of heteroatoms to Pt(II) [N1―Pt1: 0.2019(5) nm, N2―Pt1: 0.1934(5) nm,N3―Pt1: 0.2012(5) nm, Cl1―Pt1: 0.2296(2) nm] all fell in the range observed from [Pt(terpy)Cl]ClO4(0.1952−0.2003 nm)37or known Pt-terpyridyl complexes38,39. The angles and torsion angles were Pt1―N1―N2, 80.71(21)°; Pt1―N1―N3,161.42(23)°; Pt1―Cl1―N1, 99.08(17)°; N2―Pt1―N1―C5,2.3(4)°; N3―Pt1―N1―C1, −179.2(6)°; and N2―Pt1―N3―C11, 0.2(4)° from which the whole molecule could be described as approximately planar. The different stacking of[Pt(terpy-TEMPO)Cl]+were observed: the Pt1−Pt2 distance[0.35235(4) nm)] was slightly longer than that of 0.3269(1) nm in [Pt(terpy)Cl]ClO437, in which the steric effects of TEMPO probably hinder short Pt―Pt distances. In order to avoid steric repulsion, the two stacked terpyridine planes would complement each other without complete overlap (Fig.S5, Supporting Information).
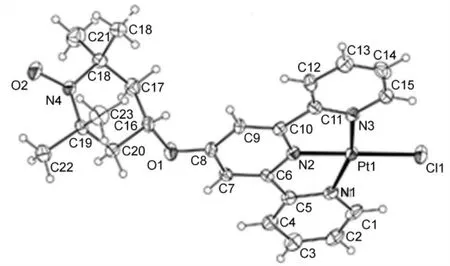
Fig.1 Perspective drawing of the complex cation of compound 2 with selected atomic numbering scheme.
3.3 UV/Vis absorption properties
As a representative example of this class of [Pt(4-Rʹ-terpy)Cl]+complexes, the electronic absorption spectra of complexes 1 and 2 in methyl alcohol are shown in Fig.2. When complexes 1 and 2 are dissolved in methyl alcohol, the absorbance of each platinum complex follows Beerʹs law and is consistent with dissolution into monomeric ions. Chargetransfer electronic absorptions of Pt(II) terpyridine complexes tend to occur in the wavelength range from 350 to 450 nm,along with mainly intraligand π−π* absorptions at shorterwavelengths40−43. On the whole, the absorption spectrum of the Pt(terpy)Cl+system of compounds 1 and 2 were very similar,naturally breaks into two energy regimes (Fig.2): broad moderate intense absorption band of 370−450 nm (band A, ε ≈103dm3·mol−1·cm−1) and an intense band with distinct vibronic structures at 270−350 nm (band B, ε > 104dm3·mol−1·cm−1).Che, and others have explored in considerable details of photophysical properties of a series of Pt(II) terpyridine derivatives44−46. Take into consideration all these situations, we tentatively assign the bands B in Fig.2 more likely to represent π−π*transitions. On the other hand, band A (ε = 3.90 × 103dm3·mol−1·cm−1) in Fig.2 is broad with moderately intensity,which makes it difficult to locate the maximum wavelength.But the band A appeared to be too low in energy to be π−π*transitions and too intense (ε ≥ 103dm3·mol−1·cm−1) to be d−d transitions, which it is most likely to be the metal to ligand charge-transfer (CT) transitions analogous to those identified in the spectra of Pt(II) bipyridine complexes47. We tentatively assign it to the metal-to-ligand charge-transfer (MLCT)transition Pt(5d) → terpy(π*). These bands of complex 1 generally shift to lower energy using methanol solvent (Table S2, Fig.S6, Supporting Information), which was consistent with a charge transfer character47,48.
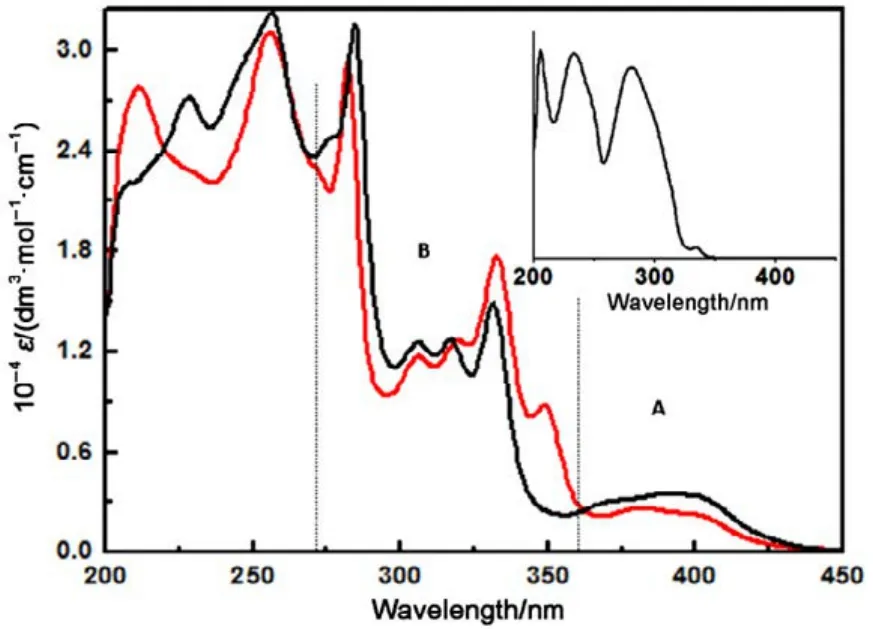
Fig.2 UV/Vis spectra of compounds 1(black) and 2(red) in methyl alcohol solvent at 298 K (color online).
3.4 Solid-State photoluminescence properties
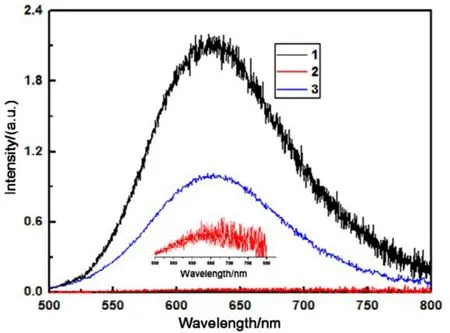
Fig.3 Emission spectra of compounds 1 (black), 2 (red), and 3 (blue) in solid state at 298 K (color online).
The entire solid samples were deoxygenated by vacuum-argon cycling during testing. Che and Gray49−51have reported the phosphorescence of the Pt(terpy)Cl+complexes at room temperature as well as 77 K in the solid. The[Pt(terpy)Cl]+complexes showed no detectable emission in its solution state at room temperature, which was due to the quenching of the3MLCT state by the thermally accessible3d−d exited state via non-radiative decay52. It was reported that the[Pt(terpy)Cl]+complexes would exhibit very strong luminescence, which was derived from Pt―Pt and/or π−π interaction, both in the solid state and in low temperature glass37. The emission spectrum of 1 ([Pt(terpy)Cl]+complex in our study, Fig.3) upon excitation at λ ≥ 400 nm displayed a strong emission in solid state at λmax630 nm at room temperature. This midrange luminescence (550−650 nm) in solid-state is more difficult to assign, although the low temperature solid-state luminescence of [Pt(terpy)Cl]X (X=PF6−, ClO4−, Cl−, CF3SO3−), ranging from 565 to 695 nm, is assigned to a singlet-singlet MMLCT (π*→ dσ*, metal-metalto ligand charge transfer, MMLCT) transition37. As the Fig.3 shows, the emission of complex 1 is a broad band, which is much too broad for an MMLCT transition in some cases18,19,and it is debatable for such a long ground-state Pt―Pt distance to assign the luminescence of complex 1 to a MMLCT transition, but excimeric ππ*MMLCT emission has been invoked to explain the very broad room-temperature emission of solid [Pt(terpy)Cl]+complexes more reasonable37. The similarly broad emission of solid [Pt(bpy)2]2+and [Pt(phen)2]2+(phen = 1,10-phenanthroline) salts also have ππ excimer character53. The excitation spectrum (Fig.S7, Supporting Information) of compound 1 was, however, complicated with unstructured, broad band (275−525 nm). Also, the complex 1 proves to be a very promising platform with an emission quantum yield of 0.17 and an excited-state lifetime of 0.94 μs(Figs.S8, S9, Supporting Information) in room-temperature solid-state. When TEMPO was pinched on the terpyridine of[Pt(terpy)Cl]+complex, the strong luminescence was quenched sharply (> 90%, Fig.3). So the complex 1 displays an intense orange emission at λmax630 nm, whereas the complex 2 displays very weak luminescence at λmax630 nm in solid state with little redshift in the emission of complex 2. Furthermore,the dramatically quenching also appeared in the excitation spectrum of complex 2, in contrast to complex 1 (Fig.S7,Supporting Information), which indicated TEMPO radical can extinguish excimeric π→π*excitation. The possible mechanisms could be interpreted as follows: The Pt(II) salts series in close proximity to each other are likely to form dimeric structure and yield efficiently the excimer emission in solid states54,55. In the case if photoexcitation of the complex 1 in solid states at room temperature with 406 nm light (hvex),complex 1 is mainly excited to form a singlet exciton1(complex 1)*(Eq.(1), which is smoothly transferred to the ground state of another1(complex 1) yield an singlet excimer1(complex 1. complex 1)*(Eq.(2)), then is transferred to the triplet3(complex 1. complex 1)*via ISC (Eq.(3)). Finally, the excimer emission (hvexcimer) is generated (Eq.(4)).

In the case of the complex 2, which modified by TEMPO,although the Pt1―Pt2 distance (0.35235(4) nm) distinguished the absence of Pt―Pt interactions, π−π interactions cannot be ignored in dimers of complex 2, and the similar process is possible when excitation occurs upon 2 to generate singlet complex 21(complex 2)*(Eq.(5). In the practical case, the phosphorescence spectra are similar between complexes 1 and 3, and therefore, the strong intermolecular interactions exist not only in complex 1, but also in complexes 2 and 3 in the solid state.1(complex 2)*contribute readily to the singlet excimer formation of complex 2 [1(complex 2. complex 2)*, Eq.(6)], and then relaxed to triplet excimer3(complex 2. complex 2)*(Eq.(7)). But the quenching due to TEMPO selectively provide the excited triplet state, so the triplet excimer [3(complex 2.complex 2)*] back to the ground state via the form of thermal radiation (Eq.(8)), thus the excimer emission cannot be engender.
In comparison with the emission spectra peaks of complexes 1 and 2, manganic effect of TEMPO on the emission wavelength was not obvious. The maximum emission wavelength of complex 2 slightly moved to ~640 nm compared with complex 1 (λem630 nm). Moreover, the luminescence lifetimes of complexes 1 and 2 were almost the same, 945 and 957 ns, respectively. Interestingly, for solid sample of complex 2, the emission lifetimes under the temperatures from 33 to 290 K were virtually the same (Figs.S10, S11; Table S3, Supporting Information). While the emission peaks of compound 2 were shifted slightly to longer wavelengths with the rising of temperature. We postulate this significant temperature independent for emission lifetimes and emission λmaxof complex 2 is consistent with excimeric ππ*excited state, which was responsible for the radiative decay of the emission.Consequently, the nitroxide radical provided efficient luminescence quenching and this radical could preferably react with ascorbic acid (Scheme 1), thus, as a result of the reaction with ascorbic acid, complex 2 became the luminous-reduced form without radical spins (complex 3, Fig.3).
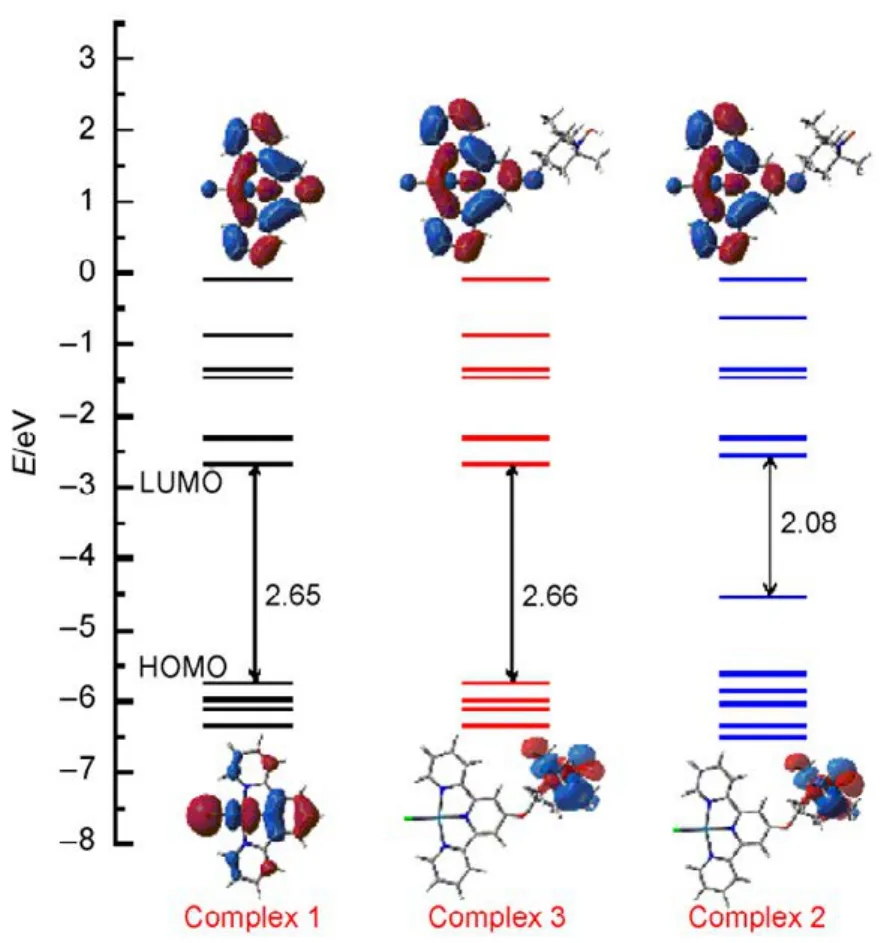
Fig.4 Diagrams of singlet states molecular orbitals,energy band gap (eV), HOMO and LUMO under TD-DFT caculations for compelxes 1, 2, 3.
The frontier molecular orbitals (FMOs) were essential to describe of the electronic and spectroscopic properties of complexes. In view of obtaining the convincible energy band gap and the energy level diagram, the HOMO and LUMO orbital of complexes 1−3 were calculated within Gaussian-09 program at hybrid B3LYP-DFT level. Full geometric optimizations were carried out using basis set 6-31+G(d,p),followed by frequency analysis to insure all local minima with real frequencies. The corresponding FMOs and energy gap (eV)were shown in Fig.4. The calculated compositions of FMOs were very different with the substituent groups (R) changed (R = H,TEMPO, TEMPOH) for Pt(II) terpyridine. The LUMO of complexes 1, 3 were almost at the same level except complex 2 gradually increased slightly, but the differences of level and distribution between three kinds of HOMO were more obvious.In Fig.4, the band gap changes in the order of 2 (2.08 eV) < 1(2.65 eV) ≈ 3 (2.66 eV), in which complex 2 possessed minimal band gap because of the nitroxide radical of terpridine.The partial frontier orbitals compositions of 1−3 were listed in Fig.4. When there was no substituent, e.g., complex 1, the HOMO orbital was mainly contributed by Cl, Pt, and pyridine.With respect to complexes 2 and 3, HOMO orbitals were localized mainly on the TEMPO or TEMPOH. And most impressively, we have noticed that the energy levels of HOMO for complex 2 raised much compared to complexes 1, 3.Obviously, the nitroxide radical (TEMPO) on the terpyridyl ligand effectively raised the energy level of HOMO and slightly lowered LUMO, thus, the energy gap between the HOMO-LUMO became the lowest among the three complexes.Accordingly, the FMOs were the intrinsic reason for the absorption and emission properties of complexes 1−3,especially the emissive performance. As it was showed in Fig.3,the luminescence of complex 1 was strong, and complex 2 decreased slightly, while the complex 3 was almost nonluminous. Hence the evidence strongly suggests that the energy levels of HOMO and LUMO were sensitive to the substituent of nitroxide radical (TEMPO), which should be responsible for the emission of complexes.
The emission intensity of complex 3 was less than that of complex 2 from the Fig.3, reasons for the difference maybe as follow: solid fluorescence intensity is related to many factors,such as the temperature, the nature of the sample itself, the amount (thickness), the integration time, the surface roughness,the degree of focus, the sample location or something else.Especially for two different samples, this difference is more significant. Therefore, we speculated this difference of fluorescence intensity in Fig.3 is caused by a combination of many factor, such as the sample roughness, the degree of micro-focus, the location and so on.
The thickness of those samples was about 10−100 μm. In addition, the luminescence property of complex 3 was similar with complex 1, which exhibit strong luminescence in the solid state and in low-temperature glass49−51, while no detectable emission was observed in their solution state at room temperature52. The potential biological applications of complex 3 is very attractive, however, those experiments need harsh experimental conditions (low temperature, no water and no oxygen). So the unstable and non-emission in solution properties of complex 3 is restrictive to its application, and our follow-up work will modify these complexes in order to apply in broader fields.
3.5 Properties of electron paramagnetic resonance spectroscopy

Fig.5 EPR spectra of T (black), L (red), and compound 2(green) in DMF at 298 K, 1 × 10−3 mol·L−1 (color online).
As the luminescence was quenched in [Pt(terpy-TEMPO)]+system, the EPR signal started to be activated. Steady-state EPR spectrum of 4-OH-TEMPO, L, and complex 2 were observed at 298 K in DMF solvent. The spectra demonstrated that fluorophores would drastically influence the rotational correlation coefficients (τR), which can be obtained via analysis of the EPR line widths and relative intensities (As detailed in literature56.57). The τR(Eq.(1), Fig.S12, Table S4, Supporting Information) of 4-OH-TEMPO, L, and complex 2 followed the order: 2.9 × 10−11s < 1.3 × 10−10s < 2.0 × 10−10s, which were all located in fast-motion region58.
As an evident difference in the Fig.5, the larger substituent group of TEMPO was introduced, the greater asymmetry of the EPR spectra of TEMPO derivatives came out. We supposed that the increasing steric hindrance of the 4-substituent groups,rotation of the TEMPO molecule becomes increasing restricted,thereby prolonging the rotational correlation time, as well as the anisotropies in the hyperfine coupling constant (A value)and g value are less effectively averaged out. Approximate calculation of rotational correlation coefficients (τR) could be obtained via analysis of the EPR line widths and relative intensities. As detailed in Ref.59, within this regime the relation between τRand the spectral parameters are given to a good approximation by the expressions in supporting information(Eq.(1), Fig.S12, Supporting Information).
4 Conclusions
In summary, we reported the synthesis and structural characterization of a novel square-planar Pt(II) complex with a terpyridine ligand decorated with TEMPO radical derivative.The complex 2 is sensitive to ascorbic acid, thus it could develop to be a luminescence and EPR bi-functional probe in detecting ascorbic acid for clarifying the biological roles.Moreover, more information about cytotoxic mechanism would be got once terpyridine Pt(II) complexes is used due to its DNA-intercalating activity60.
By the incorporation of TEMPO moiety, the3MMLCT emissive excited states of [Pt(terpy)Cl]+unit was significantly affected, leading to a new type of highly efficient quenching luminescence [Pt(terpy-TEMPO)Cl]+complex, in turn triggering the EPR signal. Last but not least, this novel[Pt(terpy-TEMPO)Cl]+complex shows promising prospect for metal-TEMPO communication and could possibly be employed as structural elements in new solid state lighting, sensing applications, or organometallic catalyst system. Simultaneously,the development of these chromophore-nitroxide sensors opened the possibilities of obtaining new “off-on”photoluminescence chemosensors, which would be designed as a kind of logic gate switch, or to monitor the process in oxidation reaction. On the analysis of the conversion of EPR signal and luminescence, more information about single electron behavior would be achieved in the process of coordination between substrates and transition metal, which could explain the TEMPO-catalyzed oxidation or cytotoxic mechanism more clearly.
The new synthetic route of [Pt(terpy-TEMPO)Cl]+could further be used to concatenate terpy-TEMPO ligand to different transient metals, generating a diverse array of transition metal-TEMPO complexes.
Acknowledgment: The authors sincerely acknowledge Dr.QIN Haiyan (Department of Chemistry, Zhejiang University,Hangzhou 310027, P. R. China) and Dr. WANG Bingjie(Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China) for helpful discussions.
Supporting Information: Details of X-ray structural analysis, experimental procedures, temperature dependence of emission spectra, UV/Vis spectra, and ESI-MS data of complexes were given. This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Ames, B. N.; Shigenaga, M. K.; Hagen, T. M. Proc. Natl. Acad. Sci.1993, 90 (17), 7915. doi: 10.1073/pnas.90.17.7915
(2) Stamler, J. S.; Singel, D. J.; Loscalzo, J. Science 1992, 258 (5090),1898. doi: 10.1126/science.128192
(3) Church, D. F. Anal. Chem. 1994, 66 (7), A418.
(4) Di, L.; Hua, Z. Adv. Synth. Catal. 2011, 353 (8), 1253.doi: 10.1002/adsc.201000876
(5) Rozantse, E.; Sholle, V. D. Synthesis. 1971, (4), 190.
(6) Dijksman, A.; Marino-Gonzalez, A.; Payeras, A. M. I. J. Am. Chem.Soc. 2001, 123 (28), 6826. doi: 10.1021/Ja0103804
(7) Karimi, B.; Biglari, A.; Clark, J. H.; Budarin, V. Angew. Chem. Int.Ed. 2007, 46 (38), 7210. doi: 10.1002/anie.200701918
(8) Ishii, K.; Takayanagi, A.; Shimizu, S.; Abe, H.; Sogawa, K.;Kobayashi, N. Free Radical Bio. Med. 2005, 38 (7), 920.doi: 10.1016/j.freeradbiomed.2004.12.017
(9) Ishii, K.; Takeuchi, S.; Shimizu, S.; Kobayashi, N. J. Am. Chem. Soc.2004, 126 (7), 2082. doi: 10.1021/Ja035352v
(10) Blough, N. V.; Simpson, D. J. J. Am. Chem. Soc. 1988, 110 (6), 1915.doi: 10.1021/Ja00214a041
(11) Ishii, K.; Hirose, Y.; Kobayashi, N. J. Phys. Chem. A 1999, 103 (13),1986. doi: 10.1021/Jp983624o
(12) Green, S. A.; Simpson, D. J.; Zhou, G.; Ho, P. S.; Blough, N. V. J.Am. Chem. Soc. 1990, 112 (20), 7337. doi: 10.1021/Ja00176a038
(13) Camerel, F.; Ziessel, R.; Donnio, B.; Bourgogne, C.; Guillon, D.;Schmutz, M.; Iacovita, C.; Bucher, J. P. Angew. Chem. Int. Ed. 2007,46 (15), 2659. doi: 10.1002/anie.200604012
(14) Tam, A. Y. Y.; Wong, K. M. C.; Wang, G. X.; Yam, V. W. W. Chem.Commun. 2007, No. 20, 2028. doi: 10.1039/B705062c
(15) Lu, W.; Law, Y. C.; Han, J.; Chui, S. S. Y.; Ma, D. L.; Zhu, N. Y.;Che, C. M. Chem. Asian J. 2008, 3 (1), 59. doi:10.1002/asia.200700265
(16) Ou, Z. Z.; Ju, B. L.; Gao, Y. Y.; Wang, Z. C.; Huang, G.; Qian, Y. M.Acta Phys. -Chim. Sin. 2015, 31 (12), 2386. [欧植泽, 句宝龙, 高云燕, 王子超, 黄 干, 钱一梦. 物理化学学报, 2015, 31 (12): 2386.]doi: 10.3866/PKU.WHXB201510137
(17) Liu, X. Y.; Han, X.; Zhang, L. P.; Tung, C. H.; Wu, L. Z. Phys. Chem.Chem. Phys. 2010, 12 (40), 13026. doi: 10.1039/c0cp00100g
(18) Bailey, J. A.; Miskowski, V. M.; Gray, H. B. Inorg. Chem. 1993, 32(4), 369. doi: 10.1021/Ic00056a001
(19) Aldridge, T. K.; Stacy, E. M. Inorg. Chem. 1994, 33 (4), 722.doi: 10.1021/Ic00082a017
(20) Lai, S. W.; Chan, M. C. W.; Cheung, K. K.; Che, C. M. Inorg. Chem.1999, 38 (19), 4262. doi: 10.1021/Ic990446k
(21) Chung, C. Y. S.; Yam, V. W. W. J. Am. Chem. Soc. 2011, 133 (46),18775. doi: 10.1021/Ja205996e
(22) Xu, P.; Wu, H. T.; Jia, H. X.; Ye, S. F.; Du, P. W. Organometallics.2014, 33 (11), 2738. doi: 10.1021/Om500115s
(23) Yam, V. W. W.; Chan, K. H. Y.; Wong, K. M. C.; Chu, B. W. K.Angew. Chem. Int. Ed. 2006, 45 (37), 6169. doi:10.1002/anie.200600962
(24) Wu, D.; Deng, K.; He, M.; Zeng, Q.; Wang, C. Chem. Phys. Chem.2007, 8 (10), 1519. doi: 10.1002/cphc.200700096
(25) Siebert, R.; Akimov, D.; Schmitt, M.; Winter, A.; Schubert, U. S.;Dietzek, B.; Popp, J. ChemPhysChem 2009, 10 (6), 910.doi: 10.1002/cphc.200800847
(26) Park, J.; Lee, J. H.; Jaworski, J.; Shinkai, S.; Jung, J. H. Inorg. Chem.2014, 53 (14), 7181. doi: 10.1021/Ic500266f
(27) Yam, V. W. W.; Tang, R. P. L.; Wong, K. M. C.; Ko, C. C.; Cheung,K. K. Inorg. Chem. 2001, 40 (3), 571. doi: 10.1021/Ic000586q
(28) Kunkely, H.; Vogler, A. J. Am. Chem. Soc. 1990, 112 (14), 5625.doi: 10.1021/Ja00170a029
(29) Connick, W. B.; Geiger, D.; Eisenberg, R. Inorg. Chem. 1999, 38(14), 3264. doi: 10.1021/Ic981387y
(30) Chan, C. W.; Cheng, L. K.; Che, C. M. Coordin. Chem. Rev. 1994,132, 87. doi: 10.1016/0010-8545(94)80027-8
(31) Tears, D. K. C.; McMillin, D. R. Coordin. Chem. Rev. 2001, 211, 195
(32) Olia, M. B. A.; Schiesser, C. H.; Taylor, M. K. Org. Biomol. Chem.2014, 12 (35), 6757. doi: 10.1039/c4ob01172d
(33) Mcdermott, J. X.; White, J. F.; Whitesides, G. M. J. Am. Chem. Soc.1976, 98 (21), 6521. doi: 10.1021/Ja00437a018
(34) Morgan, G. T.; Burstall, F. H. J. Chem. Soc. 1934, 1498.doi: 10.1039/Jr9340001498
(35) Halcrow, M. A.; Brechin, E. K.; McInnes, E. J. L.; Mabbs, F. E.;Davies, J. E. J. Chem. Soc. Dalton Trans. 1998, (15), 2477.doi: 10.1039/A803793k
(36) Price, J. H.; Schramm, R. F.; Wayland, B. B.; Williams, A. Inorg.Chem. 1972, 11 (6), 1280. doi: 10.1021/Ic50112a025
(37) Bailey, J. A.; Hill, M. G.; Marsh, R. E.; Miskowski, V. M.; Schaefer,W. P.; Gray, H. B. Inorg. Chem. 1995, 34 (18), 4591.doi: 10.1021/Ic00122a015
(38) Tang, W. S.; Lu, X. X.; Wong, K. M. C.; Yam, V. W. W. J. Mater.Chem. 2005, 15 (27−28), 2714. doi: 10.1039/B501644d
(39) Wong, K. M. C.; Tang, W. S.; Lu, X. X.; Zhu, N. Y.; Yam, V. W. W.Inorg. Chem. 2005, 44 (5), 1492. doi: 10.1021/Ic049079p
(40) McMillin, D. R.; Moore, J. J. Coordin. Chem. Rev. 2002, 229 (1−2),113. doi: 10.1016/S0010-8545(02)00041-3
(41) Yam, V. W. W.; Wong, K. M. C.; Zhu, N. Y. Angew. Chem. Int. Ed.2003, 42 (12), 1400. doi: 10.1002/anie.200390360
(42) Yam, V. W. W.; Chan, K. H. Y.; Wong, K. M. C.; Zhu, N. Y. Chem.-Eur. J. 2005, 11 (15), 4535. doi: 10.1002/chem.200500106
(43) Yu, C.; Wong, K. M. C.; Chan, K. H. Y.; Yam, V. W. W. Angew.Chem. Int. Ed. 2005, 44 (5), 791. doi: 10.1002/anie.200461261
(44) Che, C. M.; Butler, L. G.; Gray, H. B. J. Am. Chem. Soc. 1981, 103(26), 7796. doi: 10.1021/Ja00416a021
(45) Rice, S. F.; Gray, H. B. J. Am. Chem. Soc. 1983, 105 (14), 4571.doi: 10.1021/Ja00352a011
(46) Lu, W.; Chan, M. C. W.; Cheung, K. K.; Che, C. M. Organometallics 2001, 20 (12), 2477. doi: 10.1021/Om0009839
(47) Gidney, P. M.; Gillard, R. D.; Heaton, B. T. J. Chem. Soc. Dalton Trans. 1973, (2), 132. doi: 10.1039/Dt9730000132
(48) Benedix, R.; Vogler, A. Inorg. Chim. Acta 1993, 204 (2), 189.doi: 10.1016/S0020-1693(00)82924-2
(49) Yip, H. K.; Cheng, L. K.; Cheung, K. K.; Che, C. M. J. Chem. Soc.Dalton Trans. 1993, (19), 2933. doi: 10.1039/Dt9930002933
(50) Lai, S. W.; Chan, M. C. W.; Cheung, K. K.; Che, C. M.Organometallics 1999, 18 (17), 3327. doi: 10.1021/Om990256h
(51) Connick, W. B.; Henling, L. M.; Marsh, R. E.; Gray, H. B. Inorg.Chem. 1996, 35 (21), 6261. doi: 10.1021/Ic960511f
(52) Houlding, V. H.; Miskowski, V. M. Coordin. Chem. Rev. 1991, 111,145. doi: 10.1016/0010-8545(91)84019-2
(53) Miskowski, V. M.; Houlding, V. H. Inorg. Chem. 1989, 28 (8), 1529.doi: 10.1021/Ic00307a021
(54) Xu, H.; Lv, Y. F.; Zhu, W. Q.; Xu, F.; Long, L.; Yu, F. F.; Wang, Z.X.; Wei, B. J. Phys. D: Appl. Phys. 2011, 44 (41), 1.doi: 10.1088/0022-3727/44/41/415102
(55) Shigehiro, T.; Yagi, S.; Maeda, T.; Nakazumi, H.; Fujiwara, H.;Sakurai, Y. J. Phys. Chem. C 2013, 117 (1), 532.doi: 10.1021/jp307853t
(56) Robinson, B. H.; Schurr, J. M.; Kwiram, A. L.; Thomann, H.; Kim,H.; Morrobelsosa, A.; Bryson, P.; Dalton, L. R. J. Phys. Chem. 1985,89 (23), 4994. doi: 10.1021/J100269a022
(57) Thomann, H.; Cline, J. F.; Hoffman, B. M.; Kim, H.; Morrobelsosa,A.; Robinson, B. H.; Dalton, L. R. J. Phys. Chem. 1985, 89 (10),1994. doi: 10.1021/J100256a037
(58) Evans, R. G.; Wain, A. J.; Hardacre, C.; Compton, R. G. Chem. Phys.Chem. 2005, 6 (6), 1035. doi: 10.1002/cphc.200500157
(59) Arewgoda, C. M.; Bond, A. M.; Dickson, R. S.; Mann, T. F.; Moir, J.E.; Rieger, P. H.; Robinson, B. H.; Simpson, J. Organometallics.1985, 4 (6), 1077. doi: 10.1021/Om00125a022
(60) Wang, B. L.; Wang, Z. G.; Ai, F. J.; Tang, W. K.; Zhu, G. Y. J. Inorg.Biochem. 2015, 142, 118. doi: 10.1016/j.jinorgbio.2014.10.003
Synthesis, Characterization, Spectroscopic Properties, and Luminescence Quenching Mechanism of a Pt(II) Complex Decorated with a π-Conjugated TEMPO-Terpyridine Ligand System
YIN Lu1LIANG Cheng2CHEN Ke-Xian3ZHAO Chen-Xuan1YAO Jia1LI Hao-Ran1,*
(1ZJU-NHU United R&D Center, Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China;2College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, P. R. China;3School of Food Science and Biotechnology, Zhejiang Gongshang University Hangzhou, Hangzhou 310018, P. R. China)
A novel Pt(II)-based metallointercalator terpyridine complex linked with a 2,2,6,6-tetramethyl-1-piperidinyl N-oxide (TEMPO) derivative was prepared by a reaction between 4ʹ-TEMPO-terpyridine (L) and a Pt(II) salt. This complex presented unusual luminescence quenching owing to the effect of the stable nitroxide radical. The crystal structure of [Pt(terpy-TEMPO)Cl]Cl·H2O·CH3OH (terpy =2,2ʹ:6ʹ,2ʹ-terpyridine) was elucidated by X-ray crystallography. Additionally, the effect of TEMPO on the photophysical properties of [Pt(terpy-TEMPO)Cl] Cl·H2O·CH3OH was investigated by UV-Vis, fluorescence emission, and electron paramagnetic resonance (EPR) spectroscopy. Data from the absorption and luminescence properties (298 K) of the [Pt(terpy-TEMPO)Cl]+complex indicated the presence of two groups of typical bands: an intense band B with distinct vibronic structures (270−350 nm, ε > 104dm3·mol−1·cm−1) and a less intense band A (370−450 nm, ε ~103dm3·mol−1·cm−1). These two bands are generally assigned to ligand-to-ligand charge transfer (LLCT) and metal-to-ligand charge transfer (MLCT)excited states, respectively. Furthermore, efficient photoluminescent quenching behavior was observed in the emission spectra of this complex system. Quantum calculations of the molecular energy gaps and bands were performed by Gaussian 09 software. The calculated results verified that TEMPO greatly affects the energy gaps between the highest occupied molecular orbital and the lowest unoccupied molecular orbital. Thus, the relationship between efficient photoquenching and molecular structure was theoretically interpreted. EPR results indicated that when TEMPO is attached to a macrocyclic terpyridine platinum complex, e.g., [Pt(terpy)Cl]+, the terpyridine platinum complex does not affect the hyperfine coupling constant (A value) and g factor (g values) but the rotation and relaxation times of the TEMPO radical.
Terpyridine Pt(II) complex; Nitroxide radical; Synthesis; Photoluminescence;Electron paramagnetic resonance
January 11, 2017; Revised: March 17, 2017; Published online: April 11, 2017.
O641
10.3866/PKU.WHXB201704111 www.whxb.pku.edu.cn
*Corresponding author. Email: lihr@zju.edu.cn; Tel: +86-571-87952424.
The project was supported by the National Natural Science Foundation of China (21573196, J1210042), Program for Zhejiang Leading Team of S&T
Innovation (2011R50007), National High Technology Research and Development Program of China (863) (SS2015AA020601), and Fundamental Research
Funds of the Central Universities, China.
国家自然科学基金(21573196, J1210042),浙江科技创新团队项目(2011R50007),国家高技术研究发展计划项目(863) (SS2015AA020601)和中央高校基本科研业务费专项资金资助
© Editorial office of Acta Physico-Chimica Sinica
