α-MnO2纳米管自组装微球的可控制备及电化学性能
2017-11-01鞠广凯陶占良
鞠广凯 陶占良 陈 军
(南开大学化学学院,天津 300071)
α-MnO2纳米管自组装微球的可控制备及电化学性能
鞠广凯 陶占良*陈 军
(南开大学化学学院,天津 300071)
以KMnO4、HCl为反应物,H2SO4、NH4Cl为助剂,利用水热法成功合成了α-MnO2纳米管自组装微球。并采用X射线晶体衍射(XRD)、场发射扫描电子显微镜(SEM)、透射电子显微镜(TEM)以及X射线光电子能谱(XPS)表征手段对产物进行了形貌和结构表征,发现H+与Cl−离子浓度对产物的晶型有很大影响:单一增加H+或Cl−离子浓度可以使纳米管管径减小、长度增加;同时增加两种离子浓度则产物从α相转化为β相;NH4+可以起到维持产物晶型(α相)以及管状形貌的作用。电化学性能测试表明,具有独特形态的α-MnO2纳米管微球作为锂电负极具有高容量(20 mA·g−1电流密度下首周放电比容量达1783.5 mAh·g−1)与良好的倍率性能,是具有广阔应用前景的锂离子电池材料。
α-MnO2纳米管;可控制备;自组装微球;水热合成法;锂离子电池
1 引 言
过渡金属氧化物 MnO2因其物理化学特性以及价格低廉、环境友好等特点1−4,广泛应用于储能、催化、离子交换、分子吸附等领域5−8,被认为是锂电池及超级电容器等器件的理想材料。通过锰氧八面体[MnO6]基本单元的不同组合,可以形成多种晶型(如 α、β、γ、λ等)和形貌(如纳米球、纳米线、纳米立方块、纳米管等)的二氧化锰9−12,其中一维形貌的纳米管具有中空结构、更大的比表面积、更短的离子迁移路径,从而表现出良好的电化学性能13。
目前MnO2纳米管的合成方法有模板法14、水热法等15。相比较而言,模板法步骤繁琐、模板易残留,水热法因条件易控、较容易实现对MnO2晶型以及形貌控制而得到广泛应用15−17,例如较简单的一种方法是利用氧化剂 KMnO4与还原剂HCl反应制备出MnO218。由于纳米管的特殊形貌,合成难度较纳米棒、纳米线要更困难,且未对反应物离子的添加量对产物及纳米管自组装的影响加以考虑,因此探寻纳米管的合成方法及影响因素显得更有意义。
本文以 KMnO4、HCl为反应物,H2SO4、NH4Cl为助剂,采用水热法合成了 α相的 MnO2纳米管自组装微球;通过优化实验条件,固定水热时间、温度以及锰源加入量,调控了反应离子的比例,制备出不同长度与管径的纳米棒及纳米管;同时探讨了添加NH4+对产物微观形貌的影响,以及离子比例与晶型、形貌与性能之间的变化关系。
2 实验部分
2.1 α-MnO2纳米管的可控制备
α-MnO2纳米管微球采用水热法合成,反应采用优化后的温度、时间和浓度。称取 0.9482 g KMnO4(分析纯) (6 mmol)和 0.3209 g NH4Cl (分析纯)(6 mmol)溶于 24 mL 1.0 mol·L−1的 HCl和 6 mL 1.0 mol·L−1H2SO4的混合溶液中,再添加蒸馏水至总体积为70 mL。连续搅拌30 min后,将所得溶液转移到100 mL聚四氟乙烯内衬的水热釜中,在140 °C下加热18 h。收集沉淀,利用真空过滤装置过滤,在去离子水与乙醇分别洗涤三次后,80 °C下在真空烘箱中过夜烘干,记为样品6-MnO2。
为研究参与反应的离子(H+、Cl−)以及 NH4+(K+在α-MnO2晶格中有支撑孔道作用,NH4+的离子半径为0.148 nm,K+的离子半径为0.133 nm,两者离子半径接近)对产物形貌的影响,再选取 6 mmol KMnO4+ 18 mmol HCl (H+不足)、6 mmol KMnO4+ 24 mmol HCl (无添加离子)、6 mmol KMnO4+ 24 mmol HCl+ 6 mmol H2SO4(只引入H+)、6 mmol KMnO4+ 24 mmol HCl +6 mmol NH4Cl (只引入 Cl−)、6 mmol KMnO4+ 36 mmol HCl (同时引入H+与Cl−)五组反应物配比,其他反应条件不变。分别记为1-MnO2− 5-MnO2,具体见表1。
2.2 α-MnO2纳米管的表征
采用Rigaku D/max-2500 (日本)X射线晶体衍射仪 (XRD,激发光源为 Cu Kα,波长 λ ≈ 0.154 nm,管电压40 kV,管电流20 mA)对MnO2进行结构表征。通过JEOL JSM7500F场发射扫描电子显微镜(SEM)以及美国Philips Tecnai FEI透射电子显微镜(TEM)对MnO2进行形貌表征。通过日本岛津公司的Kratos Axis Ultra DLD型多功能电子能谱仪对样品进行 X射线光电子能谱(XPS)表征测定样品键合态,X射线源是Al Kα,荷电效应校正用C 1s标定。MnO2的比表面积表征利用BELSORP-mini分析仪,通过 N2等温吸-脱附曲线测得。吸附标准气体为高纯N2,分析温度为77 K,将样品预先在300 °C的氮气气氛下处理3 h。
2.3 电化学性能的测试
将制备好的 MnO2为活性材料,super-p为导电剂,PVdF(偏聚四氟乙烯)为粘结剂,质量比为7 :2 : 1并涂敷在铝箔上制成正极;隔膜采用Celgard 2320聚乙烯隔膜;负极为直径14 mm的锂片;电解液为 1 mol·L−1LiPF6的 EC/DEC 溶液(体积比为1 : 1)。以上材料在充满氩气的无水手套箱中进行电池组装。
充放电测试使用LAND测试系统,测试电压范围为 0.1−3.0 V,电流密度范围在 20−200 mA·g−1.循环伏安(CV)测试的扫描范围为0.1−3.0 V,扫速为 0.1 mV·s−1。
3 结果与讨论
3.1 XRD图谱分析
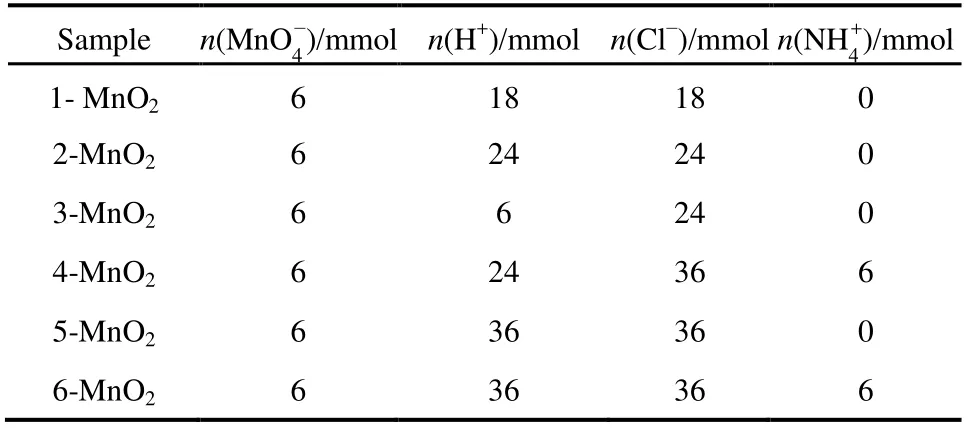
表1 不同条件下反应离子添加量Table 1 Addition of reactive ions under different conditions.
该反应利用HCl还原KMnO4制取MnO2,涉及到的化学反应方程如下:MnO+ 3Cl−+ 4H+→ α-MnO2+ 3/2Cl2+ 2H2O (1)
图1a为不同条件下制得的MnO2样品。可以看出,除5-MnO2样品为β-MnO2(a = b = 4.4041 nm,c = 2.8765 nm,P42/mnm空间群,JCPDS#81-2261)外,合成的产物均为 α-MnO2(a = b =9.7847 nm,c = 2.8630 nm,I4/m空间群,JCPDS#44-0141)。从5-MnO2生成β相MnO2,可以看出在 H+与 Cl−的共同作用下,最终产物的相发生改变;而 NH4+的加入令产物维持 α相无较大影响。H+与 Cl−共同作用对相的影响可用 Nernst方程解释:
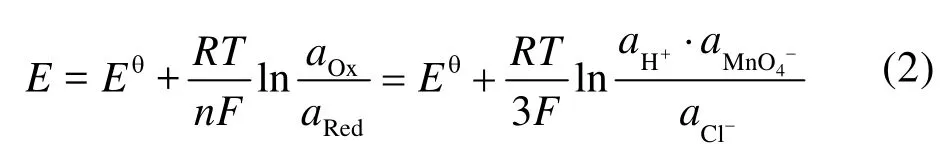
参与反应(式 1)的 H+与 Cl−浓度同时增加 1.5倍,虽然 aox与 aRed均增大,但式中[H+] : [Cl−] = 4 :3,且aox> aRed。通过计算可知反应电势差E增大,即KMnO4体现出更强的氧化能力,最终发生相的转化。同时,在水热条件的高温高压下,氧化产物Cl2的溶解度的变化对MnO2晶相同样会产生一定影响。仅增加 H+或 Cl−浓度未发生相变,进一步辅助证明 H+与 Cl−共同作用才能影响产物最终相。从反应现象上也能进一步看出反应电势的变化:水热反应进行之前,由于 MnO4−/MnO2电对(Eθ= 1.70 V)与 Cl2/Cl−电对(Eθ= 1.36 V)相差 0.34 V,在搅拌时即有晶核产生,在溶液与空气接触表面形成薄膜;加入 HCl过量的 5-MnO2形成薄膜最厚;而缺乏 H+的 1-MnO2生成薄膜速率最慢、液面形成薄膜也最少。
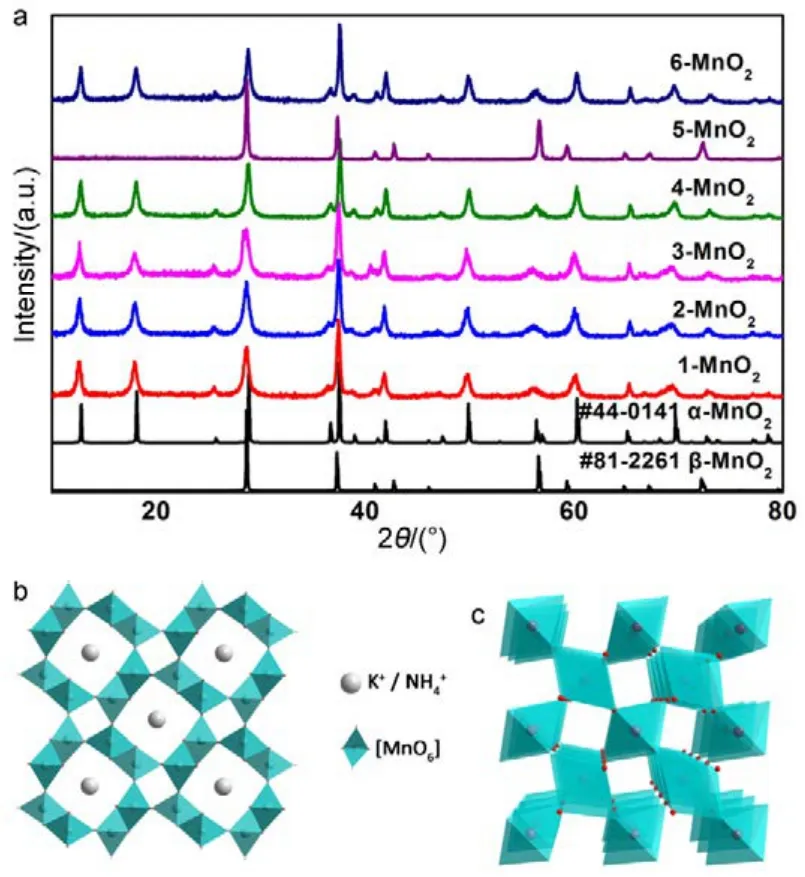
图1 (a) 不同合成条件下样品XRD衍射图,(b) α-MnO2晶体结构示意图,(c) β-MnO2晶体结构示意图Fig.1 (a) Images of XRD patterns of samples under different conditions, (b) crystal structure of α-MnO2,(c) crystal structure of β-MnO2.
3.2 电镜结果分析
图2(a1−a3)为KMnO4与HCl添加比例为1 : 3的1-MnO2电镜图像。由图2a1和图2a2的SEM照片能看出 1-MnO2呈棒状结构,分布较为零散;长度约为1.5 μm,直径150 nm左右,且部分纳米棒有分支结构,这一点也可以在图2a3的TEM图像中加以印证。TEM图像显示纳米棒未能形成中空结构。

图2 不同合成条件下MnO2样品电镜图Fig.2 Electron microscopic images of MnO2 samples under different synthesis conditions.(a1, a2) SEM images and (a3) TEM image of 1-MnO2, (b1, b2) SEM images and (b3) TEM image of 2-MnO2, (c1, c2) SEM images and (c3)TEM image of 3-MnO2, (d1, d2) SEM images and (d3) TEM image of 4-MnO2 (e1, e2) SEM images and (e3) TEM image of 5-MnO2.
图 2(b1−b3)为未额外添加离子的 2-MnO2的电镜图像。由图2b1和图2b2的扫描电镜照片可以看出,2-MnO2呈均匀分散的棒状结构,长度约为2 μm,直径约为150−200 nm,且从高倍电镜图片看出截面为有孔方形。图 2b3的透射电镜图像看出纳米棒呈中空结构,说明成功合成出 MnO2纳米管,与扫描电镜结果相一致。
图2(c1−c3)为添加H2SO4(不引入参与反应的阴离子)的3-MnO2电镜图像。由图2c1、2c2的扫描电镜可以看出,加入H+后纳米管径变小,管长增大,且产物形貌更加均一,从高倍SEM图像中可清楚看到管的截面。MnO2纳米管长度约为3 μm,管径约为70−100 nm。MnO2纳米管自发组成微球状结构,微球的直径约为5 μm。由图2c3的透射电镜表明合成出的 MnO2纳米棒同样具有孔道结构,只增大H+浓度仍可得到α-MnO2纳米管。
图 2(d1−d3)为添加 Cl−的 4-MnO2电镜照片。图2d1显示加入NH4Cl同样令纳米管自发组成球状结构。纳米管长度约为1 μm,管径为70−80 nm,截面明显变为圆形。自组装微球直径约为3 μm。由图 2d3透射电镜图片可以看出产物形貌与加入H2SO4相似,仍为中空管状结构。该样品合成时仅增大Cl−浓度,进一步表明H+与Cl−的共同作用影响最终产物晶型。
图 2(e1−e3)为同时添加 H+与 Cl−的 5-MnO2电镜图像。可以看出当KMnO4与HCl比例增至1 : 6时,产物形貌随着相转化变为双锥形,颗粒粒径约为 1 μm。
图3为添加H2SO4以及NH4Cl的6-MnO2SEM和TEM图。由图3a、3b的SEM图片可知,6-MnO2纳米管自发组成均一、分散的微球,球体直径约为5−7 μm。单体纳米管长度为5 μm左右,直径为50−70 nm。高倍扫描电镜可以明显看出截面有孔状结构。图3c的透射电镜图片进一步显示出自组装的微球结构,可知微球是由均一的 MnO2纳米管组装而成,且微球具有中空结构,可以判断产物具有较大的比表面积,从而有利于电解液的浸润与Li+离子传导。图3d的TEM图片看出MnO2的中空管状结构,管外径在50 nm左右,内径约为30 nm。图3e为HRTEM图片,显示出6-MnO2的晶面间距为0.31 nm和0.24 nm,分别对应晶体结构的(310)晶面和(211)晶面。其两晶面夹角为58°,也与四方晶系 α-MnO2的根据夹角公式计算得到理论值57.18°相接近。TEM与XRD结构表征相一致。图3f的STEM-mapping图看出Mn、O、K元素均匀分散在纳米管中,K的存在起到稳定结构的作用,利用EDS表征得到Mn与K原子个数比为9 : 1。
3.3 N2吸-脱附曲线分析
图4为6-MnO2的N2吸脱附曲线以及BJH孔径分布图。图中表面孔径分布约为2.7 nm,以介孔为主,可能属于纳米管内部孔道孔径;氮气吸脱附曲线显示为 III型曲线。利用 BET方程计算比表面积约为 27.9 m2·g−1,样品具有较大的比表面积。
3.4 XPS谱图分析

图3 6-MnO2自组装微球电镜谱图Fig.3 Electron microscopic images of 6-MnO2.(a, b) SEM images, (c, d) TEM images, (e) HRTEM image,(f) STEM-mapping.

图4 6-MnO2样品的N2吸脱附曲线Fig.4 N2 adsorption/desorption curves of 6-MnO2.
为进一步分析 6-MnO2的元素以及价态分布,进行X射线光电子能谱(XPS)测试。图5a为表面XPS全谱,Mn 2p、O 1s、K 2p峰的存在说明产物为锰氧化物,同时与mapping谱图相一致。在400 eV附近微弱的N 1s峰存在,与NH4+中N元素结合能吻合,证明产物中含有N元素;但N元素的峰面积与K元素以及Mn元素相比过小,故替换比例较小。图5b为O 1s的图谱,可以看到在529.3与531.4 eV有两个特征峰,分别对应Mn−O−Mn以及Mn−O−H两个氧化键19,与文献报道过MnO2中O的结合能相符。图5c为Mn 2p窄图谱,图中显示出Mn主要以+4价的价态存在13,同时存在少量的+3价,拟合后+4 (642.2 eV)与+3 (641.6 eV)峰面积之比约为5.2 : 1,证明Mn混合价态为+3.84。Mn 2p3/2与Mn 2p1/2主峰分别为642.0 eV与653.8 eV,相差11.6 eV,与文献报道过MnO2中Mn的结合能相吻合14。图5d为K的XPS图谱。利用峰面积计算可得Mn与K原子个数比约为7.8 : 1,与EDS分析结果相近。因此,Mn与O已发生键合作用,同时看出样品中含有 N、K元素。由于Mn存在部分+3价,除氧缺陷存在可能外,亦能说明K+、NH4+等阳离子的存在,与之前数据相一致。
3.5 MnO2自组装微球的形成机制
水热反应中相转变的过程简要说明如下。α-MnO2为2 × 2的[MnO6]孔道,在水热过程中由于受到高压以及H+与Cl−的共同影响,使得2 × 2孔道坍缩为1 × 1孔道,导致相转化的发生,对管状形貌维持有负面影响。釜内压力不变时,增加H+的量会增大纳米管长径比、管径缩小,形成均一的自组装微球(见图 2(c1, d1))。α-MnO2纳米管的形成是一个生成-化学腐蚀过程21。首先由层状的γ-MnO2晶核向四周发散形成纳米棒15,22,由于K+在晶格中的支撑,此时为α-MnO2的2 × 2孔道结构。待纳米棒生长到一定程度时,由于两极面表面能更低,H+对纳米棒两端进行腐蚀,直至完全形成中空结构,与此同时纳米管继续沿径向生长18。从图2(a3, b3, c3)的透射图中可以看出,纳米管中空结构的形成呈递进态势,中空结构从无到有,且不断增大;同时从纳米管不规则断面形状亦可证明腐蚀机制。当 H+与 Cl−浓度同时增大时,层状γ-MnO2晶核直接坍塌为1 × 1孔道结构β-MnO2。但加入NH4+后晶相形貌仍为α-MnO2纳米管微球,推测NH4+与K+离子半径接近(NH4+的离子半径为0.148 nm,K+的离子半径为0.133 nm),同样可以进入晶格起到支撑的作用,从而抑制了H+与Cl−对晶相的影响。从表2数据看出,H+浓度的增大也加速了对纳米棒的生长以及腐蚀,使得产物管长变长、管径变细,并且维持晶核反应开始时四周发散的趋势,形成球状。由图 3中透射图像也能看出 MnO2纳米管底部相连组成微球,进一步说明是由同一晶核生长而来。
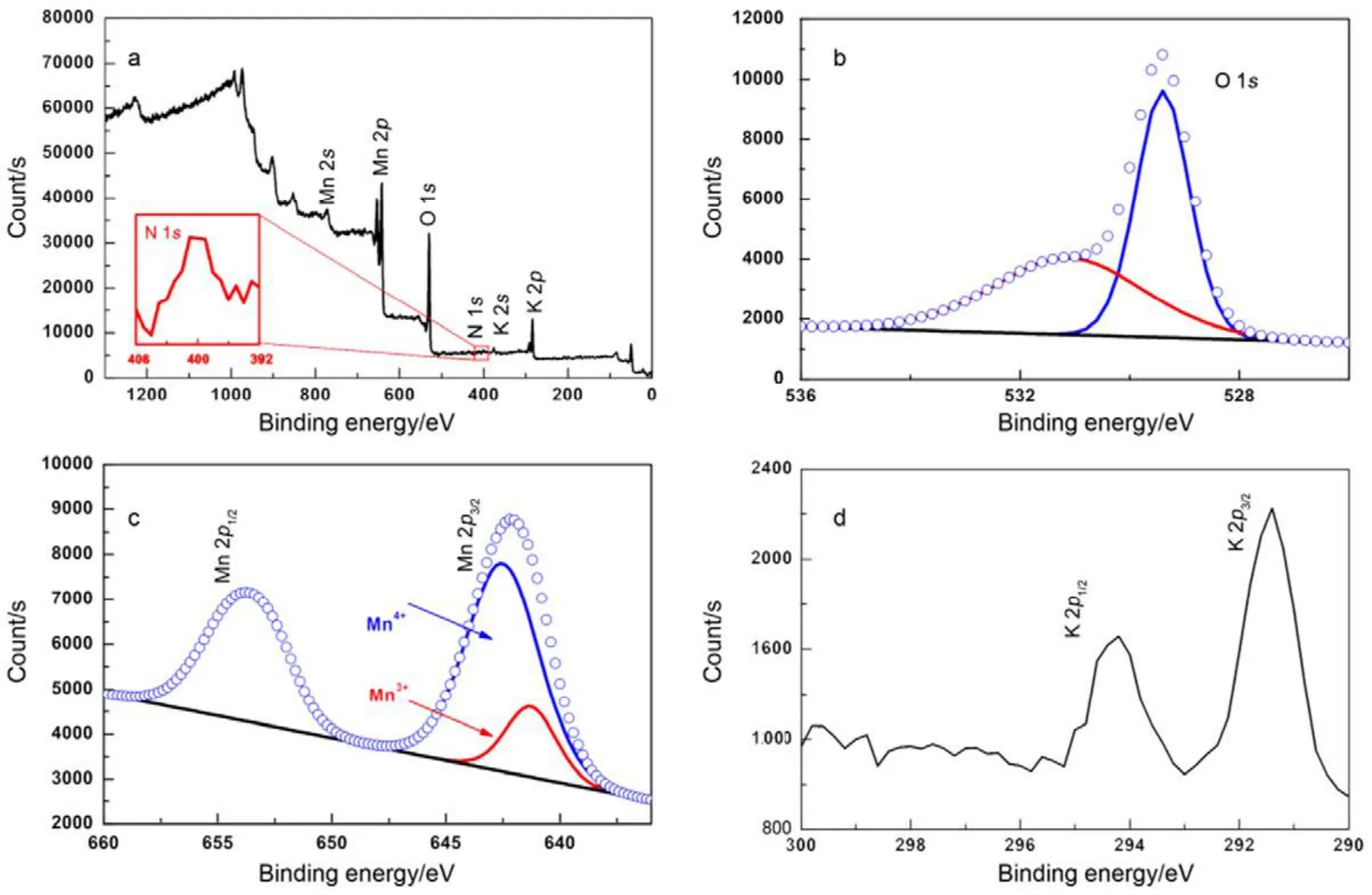
图5 6-MnO2样品XPS谱图Fig.5 XPS spectra images of 6-MnO2.(a) Surface scanning XPS spectra of 6-MnO2, (b) O 1s, (c) Mn 2p and (d) K 2p core level XPS spectra of 6-MnO2.

表2 不同管/棒状样品形貌参数对比Table 2 Comparison of morphology by samples.
3.6 MnO2自组装微球电化学测试及循环前后形貌表征
图6a为6-MnO2的CV图像,可以看出首周在1.3 V左右有一个宽峰,这对应着首周SEI膜的生成。首周在0.2 V的还原峰在第二周、第三周变为约0.25 V;同时首周在0.5 V宽化的还原峰在第二周亦移至0.4 V左右,说明循环第二周后极化减小,SEI膜亦趋于稳定。

图6 6-MnO2的电化学曲线Fig.6 Electrochemical curves of MnO2.

图7 6-MnO2样品循环前后的扫描电镜图Fig.7 SEM image of 6-MnO2 (a) before and(b) after charge-discharge cycles.
图 6b 为 6-MnO2在 100 mA·g−1电流密度下的循环以及库伦效率图,可以看出库伦效率稳定在100%左右。首周放电比容量达到 1608 mAh·g−1(理论容量为 1233 mAh·g−1),与文献对比(100、~1400 mAh·g−1)23可看出自组装微球具有更高的首周比容量。但首周 SEI膜形成,造成很大的不可逆容量,第二周变为954 mAh·g−1。容量衰减可能由于纳米材料表面 SEI膜形成以及凝胶化膜生成24,从而造成容量变化,相似现象在过渡金属氧化物(如 CoO、NiO、Fe2O3等)均有报道25。
图6c为6-MnO2微球的充放电曲线,主要放电平台约为0.5 V。在20 mA·g−1的电流密度下,首周容量可以放出 1783.5 mAh·g−1的比容量,显示出纳米微球高容量的特点。但第二周容量即减小到1276 mAh·g−1,同时1.3 V左右的放电平台消失,这与CV曲线证明的1.3 V为SEI膜生成相吻合。
图 6d为 6-MnO2的倍率放电容量曲线。将6-MnO2分别以 20、50、100 和 200 mA·g−1的电流密度进行充放电,结果表明:6-MnO2表现出较好的倍率性能,第5周的放电比容量为873 mAh·g−1,在第35周恢复到750 mAh·g−1以上。较大的比容量与良好的倍率性能归功于 6-MnO2纳米微球特殊的微纳结构,大的比表面积使电极材料与电解液充分接触,从而大大提高首周容量。但同时也造成副反应增加,导致第二周的比容量有较严重的衰减。组成微球的纳米管具有中空介孔结构,从而缩短离子扩散路径,有利于锂离子在材料中的快速脱嵌;形成的管-微球分级结构也有利于降低内部电阻26−28。
为了考察α-MnO2纳米微球的结构稳定性,对材料循环前后形貌加以表征。图7a为6-MnO2材料充放电循环前的SEM图,可以看出清晰的棒状结构以及小颗粒的导电碳。在 100mA·g−1电流密度下循环20周后,将电极片清洗后进行电镜观察,电极上仍可清晰看出棒状结构(图 7b),证明循环后形貌得到保持,但表面变得凹凸不平,这主要是循环过程中SEI膜造成的。
4 结 论
本文通过简单的水热合成法,采用 HCl与KMnO4为反应物,通过调节其比例制备出自组装α-MnO2纳米管微球。在HCl与KMnO4为4 : 1的反应体系中形成四方状纳米管,单一加入过量的H+或Cl−均可形成均一稳定的纳米管微球,同时过量则会生成 β-MnO2;而此时再加入 NH4+产物仍形成纳米管微球。证明了 H+与 Cl−共同作用对相的影响,以及NH4+在晶格中的支撑作用。在锂电池电化学测试中,纳米管微球表现出高容量(20 mA·g−1电流密度下首周放电比容量达 1783.5 mAh·g−1)与较好的倍率性能,这主要得益于纳米管微球具有球状结构中大的比表面积与管状结构中更短的离子穿梭路径。该方法制备的二氧化锰电极材料有望在锂离子电池及其他电池体系中得到应用。
(1) Zhang, K.; Han, X.; Hu, Z.; Zhang, X.; Tao, Z.; Chen, J. Chem. Soc.Rev. 2015, 44, 699. doi: 10.1039/c4cs00218k
(2) Chen, J.; Wu, F. Appl. Phys. A 2004, 78, 989.doi: 10.1007/s00339-003-2419-7
(3) Zhu, Z. Q.; Cheng, F. Y.; Chen, J. J. Mater. Chem. A 2013, 1, 9484.doi: 10.1039/c3ta00114h
(4) Cai, Z.; Xu, L.; Yan, M.; Han, C.; He, L.; Hercule, K. M.; Niu, C.;Yuan, Z.; Xu, W.; Qu, L.; Zhao, K; Mai, L. Nano Lett. 2015, 15, 738.doi: 10.1021/nl504427d
(5) Wang, L. J.; Zhang, K.; Hu, Z.; Duan, W. C.; Cheng, F. Y.; Chen, J.Nano Research 2014, 7, 199. doi: 10.1007/s12274-013-0387-6
(6) Wang, D.; Zhao, Y.; Xu, X.; Hercule, K. M.; Yan M.; An, Q.; Tian,X.; Xu, J.; Qu, L.; Mai, L. Nanoscale 2014, 6, 8124.doi: 10.1039/c4nr01941e
(7) Chae, E.; Gim, J.; Song, J.; Kim, S.; Mathew, V.; Han, J.; Boo, S.;Kim, J. RSC Adv. 2013, 3, 26328. doi: 10.1039/c3ra42897d
(8) Choi, N. S.; Chen, Z.; Freunberger, S. A.; Ji, X.; Sun, Y. K.; Amine,K.; Yushin, G.; Nazar, L. F.; Cho, J.; Bruce, P. G. Angew. Chem. Int.Ed. 2012, 51, 9994. doi: 10.1002/anie.201201429
(9) Kuo, Y. L.; Wu, C. C.; Chang, W. S.; Yang, C. R.; Chou, H. L.Electrochim. Acta 2015, 176, 1324.doi: 10.1016/j.electacta.2015.07.151
(10) Zhang, Q. X.; Peng, D.; Huang, X. J. Electrochem. Commun. 2013,34, 270. doi: 10.1016/j.elecom.2013.07.005
(11) Si, P.; Chen, P. ; Kim, D. H. J. Mater. Chem. B 2013, 1, 2696.doi: 10.1039/c3tb20341g
(12) Hu, X.; Han, X.; Hu, Y. X.; Cheng, F. Y.; Chen, J. Nanoscale 2014,6, 3522. doi: 10.1039/c3nr06361e
(13) Zhan, D.; Zhang, Q.; Hu, X.; Peng, T. RSC Adv. 2013, 3, 5141.doi: 10.1039/c3ra23258a
(14) Li, H.; Wang, W.; Pan, F.; Xin, X.; Chang, Q.; Hu, X. Mater. Sci.Eng. B-Adv. 2011, 176, 1054. doi: 10.1016/j.mseb.2011.05.041
(15) Dai, Y.; Li, J. H.; Peng, Y.; Tang, X. F. Acta Phys. -Chim. Sin. 2012,28 1771. [戴 韵, 李俊华, 彭 悦, 唐幸福. 物理化学学报, 2012,28, 1771.] doi: 10.3866/PKU.WHXB201204175
(16) Chen, J.; Cheng, F. Y. Accounts Chem. Res. 2009, 42, 713.doi: 10.1021/ar800229g.
(17) Grote, F.; Kuehnel, R. S.; Balducci, A.; Lei, Y. Appl. Phys. Lett.2014, 104. 053904. doi: 10.1063/1.4864285
(18) Luo, J.; Zhu, H. T.; Fan, H. M.; Liang, J. K.; Shi, H. L.; Rao, G. H.;Li, J. B.; Du, Z. M.; Shen, Z. X. J. Phys. Chem. C 2008, 112, 12594.doi: 10.1021/jp8052967
(19) Matsunaga, T.; Komatsu, H.; Shimoda, K.; Minato, T.; Yonemura,M.; Kamiyama, T.; Kobayash, S.; Kato, T.; Hirayama, T.; Ikuhara,Y.; Arai, H.; Ukyo, Y.; Uchimoto, Y.; Ogumi, Z. Chem. Mater. 2016,28, 4143. doi: 10.1021/acs.chemmater.5b05041
(20) Dong, S.; Chen, X.; Gu, L.; Zhou, X.; Li, L.; Liu, Z.; Han, P.; Xu, H.;Yao, J.; Wang, H.; Zhang, X.; Shang, C.; Cui, G.; Chen, L. Energy Environ. Sci. 2011, 4, 3502. doi: 10.1039/c1ee01399h
(21) Sinha, A. K.; Basu, M.; Pradhan, M.; Sarkar, S.; Negishi, Y.; Pal, T.J. Phys. Chem. C 2010, 114, 21173. doi: 10.1021/jp107757f
(22) Cheng, F. Y.; Su, Y.; Liang, J.; Tao, Z.; Chen, J. Chem. Mater. 2010,22, 898. doi: 10.1021/cm901698s
(23) Lai, H.; Li, J.; Chen, Z. ; Huang, Z. ACS Appl. Mater. Interfaces 2012, 4, 2325. doi: 10.1021/am300378w
(24) Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J. M.Nature 2000, 407, 496. doi: 10.1038/35035045
(25) Li, J.; Xi, B.; Zhu, Y.; Li, Q.; Yan, Y.; Qian, Y. J. Alloy. Compd.2011, 509, 9542. doi: 10.1016/j.jallcom.2011.07.064
(26) Zhang, X.; Cheng, F. Y.; Yang, J.; Chen, J. Nano Lett. 2013, 13,2822. doi: 10.1021/nl401072x
(27) Zhang, X. X;. Ran, F.; Fan, H. L.; Kong, L. B.; Kang, L. Acta Phys. -Chim. Sin. 2014, 30, 881. [张宣宣, 冉 奋, 范会利, 孔令斌,康 龙. 物理化学学报, 2014, 30, 881.]doi: 10.3866/PKU.WHXB201403061
(28) Xiang, X. D.; Lu, Y. Y.; Chen, J. Acta Chim. Sin. 2017, 75, 154.[向兴德, 卢艳莹, 陈 军. 化学学报, 2017, 75, 154.]doi: 10.6023/A16060275
Controllable Preparation and Electrochemical Performance of Self-assembled Microspheres of α-MnO2Nanotubes
JU Guang-Kai TAO Zhan-Liang* CHEN Jun
(College of Chemistry, Nankai University, Tianjin 300071, P. R. China)
Self-assembled microspheres of α-MnO2nanotubes were successfully synthesized by hydrothermal method using KMnO4and HCl as reactants, and H2SO4and NH4Cl as auxiliaries. X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) were used to characterize the structure and morphology of the products. The H+and Cl−ion concentrations substantially influence the crystal form of the product.Increasing either H+or Cl−ion concentration decreases the diameter of nanotubes but increases their length. In contrast, increasing both H+and Cl−ion concentrations, changes the product from α phase to β phase. Moreover, NH4+ion plays the key role of maintaining the product crystal and its tubular morphology.The electrochemical performance results showed that the microspheres of α-MnO2nanotubes with a unique morphology have a high first cycle discharge capacity of 1783.5 mAh·g−1at the current density of 20 mA·g−1, along with a good rate performance. This suggests that the self-assembled microspheres were a promising material for lithium-ion batteries.
α-MnO2nanotubes; Controllable preparation; Self-assembled microspheres;Hydrothermal synthesis; Lithium-ion battery
March 1, 2017; Revised: March 24, 2017; Published online: April 7, 2017.
O646;O614.71
10.3866/PKU.WHXB201704077 www.whxb.pku.edu.cn
*Corresponding author. Email: taozhl@nankai.edu.cn; Tel: +86-22-23504482.
The project was supported by the National Key R&D Program of China (2016YFB0901502) and the National Natural Science Foundation of China (21231005,51371100)
国家重点研发计划(2016YFB0901502)和国家自然科学基金(21231005, 51371100)资助项目
© Editorial office of Acta Physico-Chimica Sinica
