3,4,5-三甲氧基查尔酮的结构修饰与抗肿瘤活性研究进展
2017-11-01韩潇
韩 潇
(长江职业学院,湖北 武汉 430074)
3,4,5-三甲氧基查尔酮的结构修饰与抗肿瘤活性研究进展
韩 潇
(长江职业学院,湖北 武汉 430074)
查尔酮是一类广泛存在于自然界中的黄酮类化合物,具有抗肿瘤、抗菌、抗病毒等多种生物活性。近年来,通过对查尔酮进行大量的结构修饰,发现3,4,5-三甲氧基查尔酮是一类具有良好的抗肿瘤活性的新型查尔酮。本文主要对3,4,5-三甲氧基查尔酮的结构修饰及抗肿瘤作用的研究进展进行综述,旨在为今后开发3,4,5-三甲氧基查尔酮类抗肿瘤药物提供依据。
3,4,5-三甲氧基查尔酮;结构修饰;抗肿瘤活性
查尔酮是一类具有1,3-二苯基丙烯酮骨架的化合物,属于黄酮类化合物。它们广泛分布在自然界药物植物红花、甘草、镰形棘豆等植物中[1]。查尔酮的α, β-不饱和酮有较大的柔韧性,能够与众多的受体结合,产生多种生物学活性。作为植物体内黄酮合成的前体,查尔酮本身也具有广泛的生物活性,如抗肿瘤、抗真菌、抗溃疡、抗疟疾、抗炎、抗氧化等[2],而对其抗肿瘤活性的研究更为广泛。查尔酮不仅能够抑制多种肿瘤细胞的形成、增殖,还能诱导肿瘤的分化和凋亡,有望成为一类高效低毒的天然新型抗肿瘤药物。然而,查尔酮自身作用强度较弱,限制了其在临床上的应用。因此,对查尔酮进行结构修饰,以期得到抗肿瘤活性更高的衍生物具有重要的意义。事实上,目前已经数百种具有良好抗肿瘤活性的查尔酮衍生物被设计、合成,尤其是具有3,4,5-三甲氧基的查尔酮展现出了更强的抗癌活性。近年来,国内外的研究者们对3,4,5-三甲氧基的查尔酮也进行了大量的修饰,以提高其抗癌活性。从结构上看,3,4,5-三甲氧基的查尔酮也是一类1,3-二苯基丙烯酮(图1),人们对其进行结构修饰主要是集中在芳香环和α, β-不饱和酮上,由此获得了一些具有高活性的查尔酮衍生物。本文主要对近年来3,4,5-三甲氧基类查尔酮的结构修饰及构效关系的研究进展进行综述,旨在为开发3,4,5-三甲氧基类查尔酮药物提供参考。
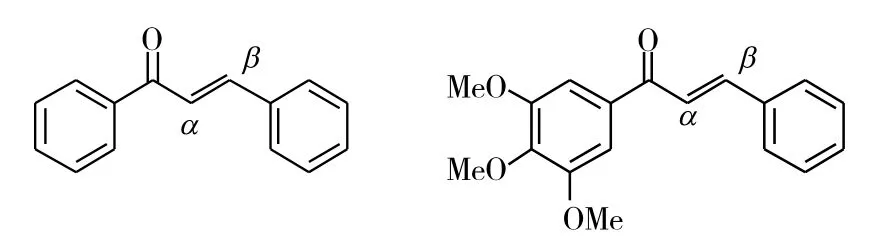
图1 查尔酮的基本结构
1 苯环修饰
考布他汀A4 (图2,CA-4)是临床上抑制微管蛋白聚合的抗肿瘤药物,从其结构分析发现,该药物具有3,4,5-三甲氧基团,对其进行结构修饰后发现,3,4,5-三甲氧基是一个抗肿瘤药效团。随后,Rao等[3]在保留了3,4,5-三甲基的活性基团的基础上,合成了一系列的3,4,5-三甲基查尔酮 (化合物1),这些查尔酮对Jurkat、U937、PBMCs肿瘤细胞均有抑制作用,其中含2-OH的化合物2对Jurkat和U937展现出了最强的抑制活性,其IC50分别是1.7μM、1.5μM。为了进一步探讨3,4,5-三甲基对抗肿瘤活性的影响,Shenvi等[4]将3,4,5-三甲基修饰成2,4,5-三甲基,合成了一系列的查尔酮3。这些查尔酮对Hela、SW-982和MCF-7均仅展现出了中等的抑制活性,其中含有-NO2的查尔酮4(IC50=16.4μM)对乳腺癌有最强的抑制活性,但仍低于临床上应用的他莫昔芬(IC50=15.6μM)。然而,进一步将这些查尔酮修饰成黄酮(化合物5)或二氢黄酮(化合物6)后,均显著降低了抗肿瘤活性,部分化合物甚至丧失了抗肿瘤活性。这些结果进一步说明,3,4,5-三甲基是一个抗肿瘤药效团,改变其甲氧基的位置将降低抗癌活性。

图2 化合物CA-4和化合物1~6的化学结构
2 α-取代修饰
Ducki等[5]首先在查尔酮的双键α位引入甲基,得到的化合物7(IC50=0.21nM)是一个具有纳摩尔抗肿瘤活性的化合物。接着,将氟、氰基和酯基引入了双键α位(化合物8~10),但是与甲基相比,并没有显著提高抗肿瘤活性。尽管化合物7具有较强的活性,然而该化合物也存在着生物利用度较差、稳定性差和溶解性差等缺点,使得该化合物未能应用于临床。因此开发出有较高的生物利用度、较好的稳定性和溶解性的查尔酮,仍然是一个重要的课题。但是在后续的研究中3,4,5-三甲氧基这一活性基团被保留着。
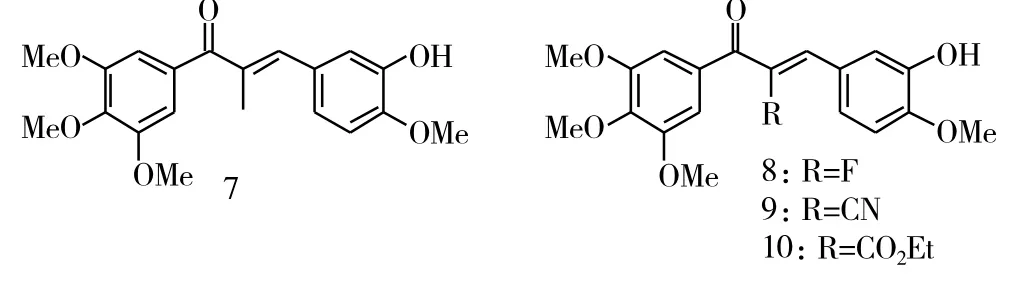
图3 化合物7~10的化学结构
3 羰基修饰
3,4,5-三甲氧基查尔酮具有α,β-不饱和酮结构,酮基能够参与缩合等反应来构建一系列杂环化合物。
Lee等[6]保留了3,4,5-三甲氧基苯基活性基团,将α,β-不饱和酮与硫脲反应,得到了一系列含硫脲的六元杂环类查尔酮衍生物,其中化合物11(IC50=6.3μM)对黑色素瘤细胞B16的活性较好,而化合物12(IC50=0.5 μM)则对淋巴白血病L1210有着较好的抑制活性。同时将3,4,5-三甲氧基修饰成2,5-二甲氧基的硫脲化合物也有着较好的活性,其中化合物13抗黑色素瘤细胞B16(IC50=6.1μM)的活性和抑制淋巴白血病L1210(IC50=0.4 μM)的活性,甚至略高于3,4,5-三甲氧基类化合物11和化合物12。
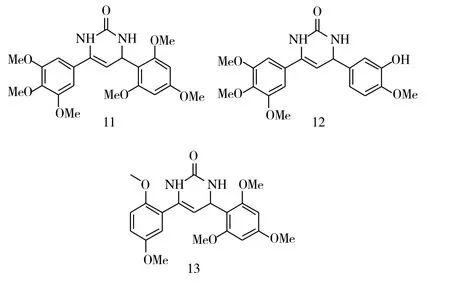
图4 化合物11~13的化学结构
将3,4,5-三甲氧基查尔酮与肼反应可形成吡唑衍生物(化合物14,图5)。这些吡唑类化合物也具有广泛的抗肿瘤活性,其中化合物15对HT-29、HCT-15、SW-620、A-549、Hop-62、HepG2、SiHa、PC-3细胞有较强的抑制活性,其IC50分别为0.47 μM、0.25 μM、0.23μM、0.87 μM、0.93 μM、8.41 μM、3.63 μM、2.64 μM。然而有意思的是,将化合物15继续修饰,即将氨基乙酰化得到化合物16后,活性明显降低,甚至丧失对部分肿瘤细胞的抑制活性,说明游离的氨基对活性影响甚大。但是,用双氧水(H2O2)将α,β-不饱和酮进行环氧化,得到的含氧三元环的查尔酮类化合物,对黑色素瘤B16和淋巴白血病L1210有着较好的抑制作用,其中化合物17(IC50=25μM)对黑色素瘤B16的抑制活性最强,化合物18(IC50=3.9μM)对淋巴白血病L1210的抑制活性最强。将这类含氧三元环化合物继续修饰,即与肼类化合物反应,也能得到吡唑衍生物,然而它们的抗肿瘤活性明显降低,部分化合物甚至丧失了抗肿瘤活性。
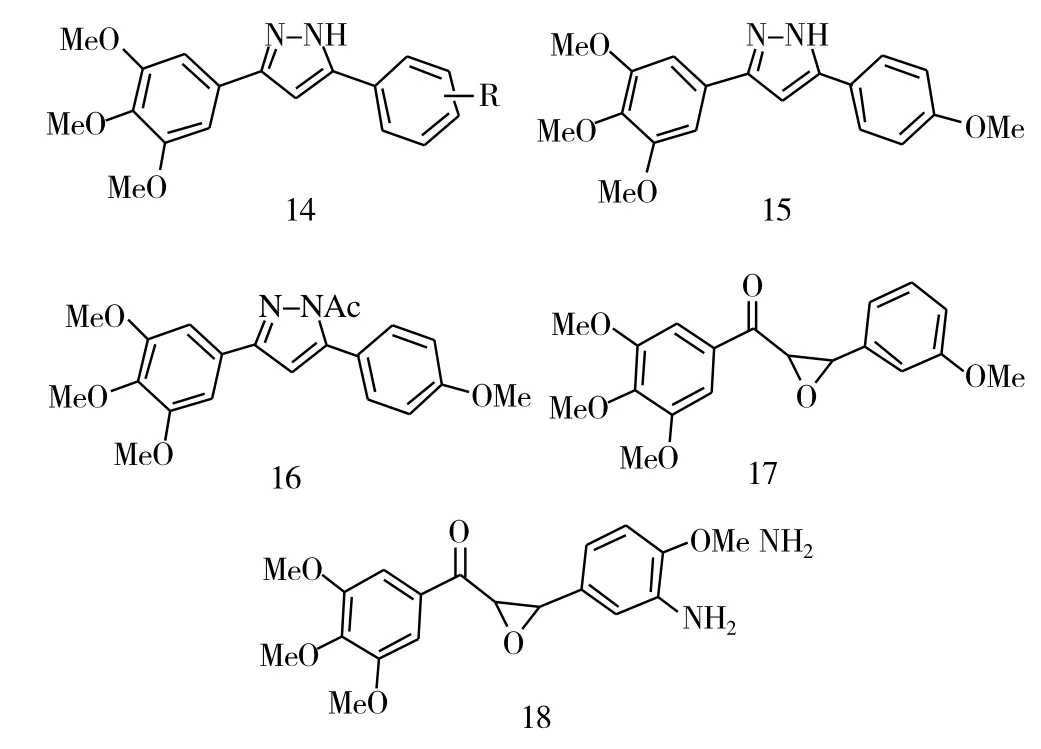
图5 化合物14~18的化学结构
4 甾体修饰
雌激素受体与乳腺癌的发生、发展和转移有着密切的联系,而甾体激素类拮抗剂能够与内源性的雌二醇竞争性地结合雌激素受体,展现出抗乳腺癌活性,已成为目前乳腺癌临床治疗的有效方法。Saxena等[7]根据药物拼接原理,将3,4,5-三甲氧基活性基团结构拼接到雌二醇上,得到了具有抗乳腺癌活性的化合物19(IC50=177.8μM)。此后以化合物19为先导化合物,继续进行结构修饰,得到了化合物20(IC50=7.3μM)。即将3,4,5-三甲氧基修饰成3,4-二甲氧基,化合物20的活性较先导化合物19提高了24倍。将化合物23的α, β-不饱和酮进行环化,得到化合物21(图6,IC50=9.88 μM),该化合物的活性与化合物7(图3)的活性相当。
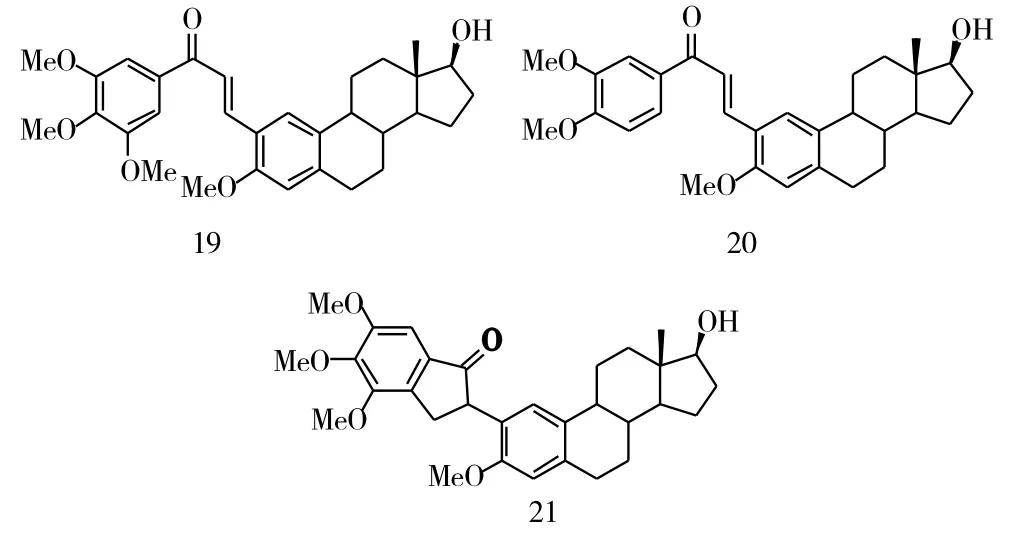
图6 化合物19~21的化学结构
5 苯并杂环修饰
苯并杂环基团也是一类抗肿瘤的药效团。为了得到新型、高效的抗肿瘤药物,将3,4,5-三甲氧基直接拼接到苯并杂环上,得到了一系列新型结构的查尔酮。Liou等[8]将3,4,5-三甲氧基拼接到吲哚上,得到了一系列的吲哚类查尔酮,大多数的化合物都对MCF-7有着较好的抑制作用,其中化合物22(IC50=1 nM)抗乳腺癌的活性最强,是化合物CA-4的8倍。但是,将羰基连接键改为硫或者磺酸酯,直接相连得到的类似物对抗乳腺癌的活性并没有提高。而将3,4,5-三甲氧基拼接到吲哚上后(化合物23~24,图7),降低了对人肺腺癌细胞A549的抑制活性。除了吲哚杂环外,将3,4,5-三甲氧基拼接到苯并噻吩上也被研究了,这类化合物从结构上看类似于雷洛昔芬,其中化合物25(GI50=3.31μM)展现了较强的抗乳腺癌活性,化合物26对HeLa、A549、HL-60、Jurkat和K562的抑制活性最强,该化合物的 IC50分别为 0.7 μM、1.4 μM、0.5 μM、0.3 μM、0.2 μM。此外,采用一锅法合成了呋喃的类似物,并对MCF-7进行了抑制的生物活性测试,其中化合物27(IC50=34 nM)的抗乳腺癌活性最高,然而与CA-4相比,化合物27的抗乳腺癌活性降低了3倍。将3,4,5-三甲氧基拼接到苯并环戊烯上,得到了一些化合物,在抗乳癌的活性研究中发现,这些化合物对MCF-7基本上没有抑制作用。此外,这类化合物对微管蛋白的抑制活性也明显低于CA-4。在这些化合物中,化合物28(IC50=36 μM)对微管蛋白的抑制活性最高,但是与CA-4(IC50=2.0 μM)相比,化合物28抗微管蛋白的活性降低了近18倍。总的来说,将3,4,5-三甲氧基拼接到不同的杂环或环戊烯上对活性有明显的影响,将3,4,5-三甲氧基拼接到吲哚环上,能够增加其生物活性,而拼接到环戊二烯上则明显降低甚至丧失了活性。这些研究为继续开发3,4,5-三甲氧基查尔酮提供了重要的依据。
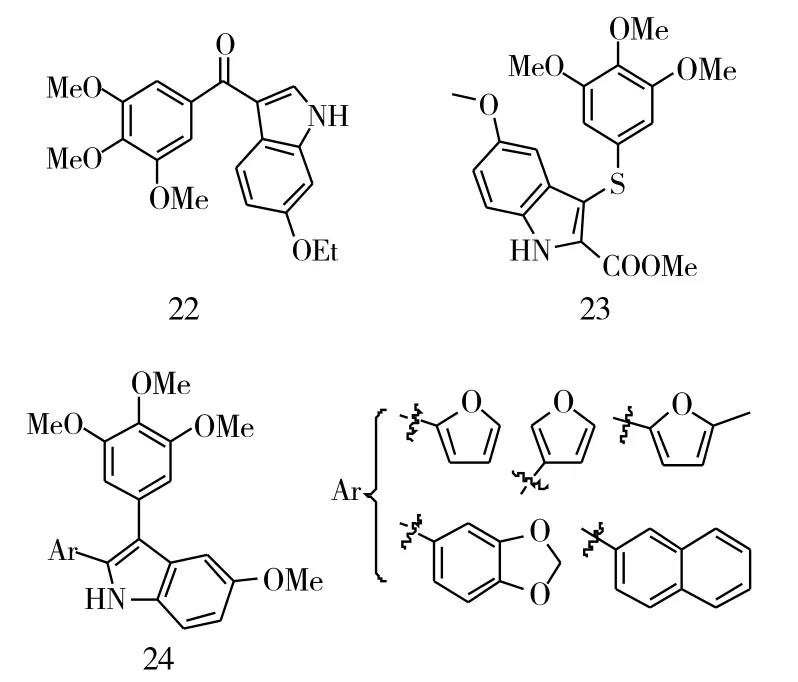
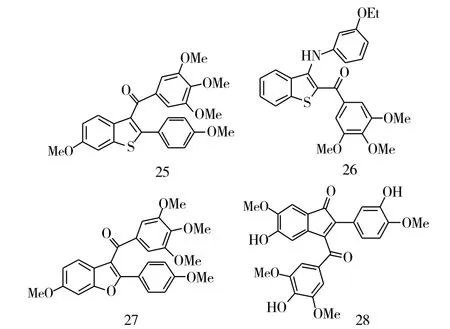
图7 化合物22~28的化学结构
6 酰胺修饰
为了提高查尔酮的水溶性,在查尔酮结构中引入了氨基。Kamal等[9]将鬼臼毒素通过酰胺键连接到查尔酮上,得到了一系列的鬼臼毒素-查尔酮的缀合物29(图8,IC50=5.3~26.7μM)。这些缀合物对多种肿瘤细胞都展现了中等的抑制活性,其中含有3,4,5-三甲基活性片段的缀合物30、31,对人肺细胞A549的抑制活性最强,它们的IC50值分别为2.7μM和2.1μM。
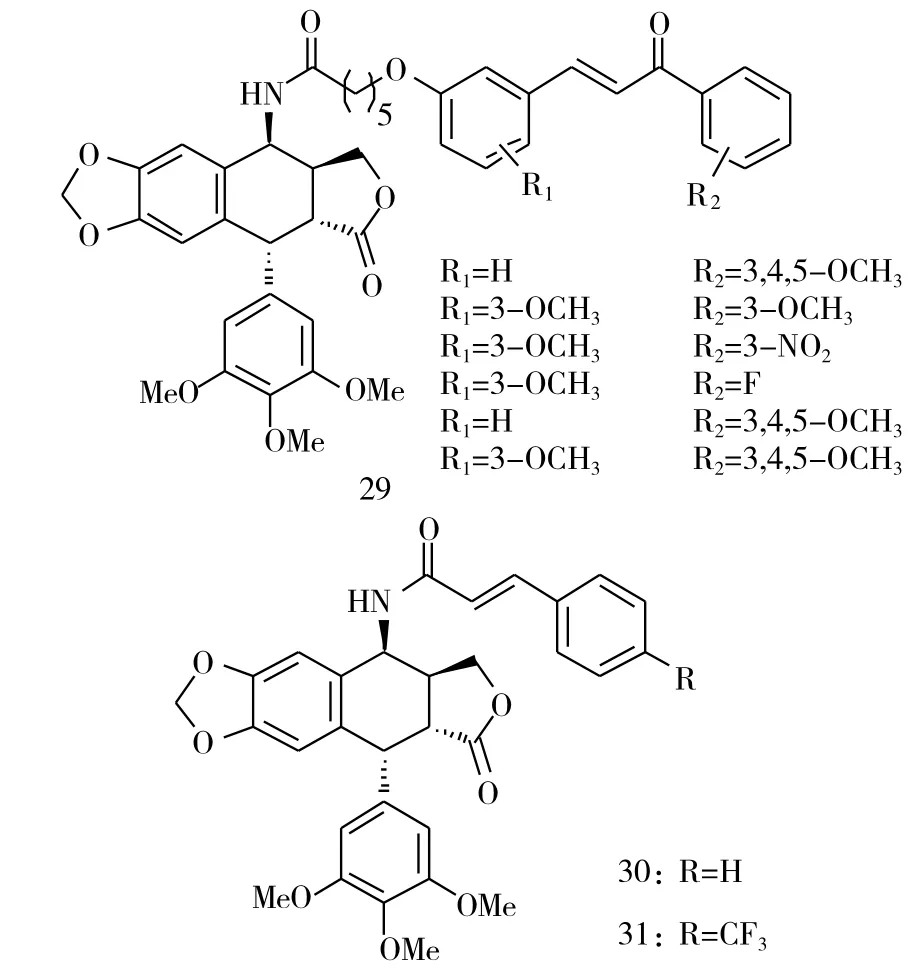
图8 化合物29~31的化学结构
将丙烯酸与3,4,5-三甲基苯胺反应,也构建了一类酰胺类的查尔酮。生物活性表明,这些酰胺查尔酮对U-937和Hela有着良好的抑制活性,其中化合物32对这2种肿瘤细胞的抑制活性最强,其IC50分别是6.0 μM和2.0 μM。有意思的是,将这类酰胺查尔酮继续进行结构修饰得到二氢查尔酮,即将α, β-不饱和酮的双键还原成单键,这些二氢查尔酮(图8,化合物33)对U-937和Hela的抑制活性显著降低,甚至丧失了活性。但是,将乙炔基换成甲氧基后,这类酰胺类的查尔酮比三苯基类化合物具有更强的抗肿瘤活性,其中化合物34对U-937的抗肿瘤活性最强。进一步改变酰胺键的位置,也能保留较强的抗肿瘤活性,其中化合物35(IC50=0.57 μM)对K562的抑制活性最强。将化合物34的邻位酰胺继续进行修饰,得到一系列的六元环类化合物(图8,化合物36),但是这类六元环类化合物的抗肿瘤活性显著低于先导化合物。此外,酰胺键的位置也被探讨了,主要是将化合物的邻位上的酰胺基团进行了修饰,得到了一系列的羧酸、酯和酰胺化合物,活性研究表明,酯也有着较强的抗肿瘤活性。在另一个苯环上引入甲氧基,对活性的影响也较大,其中3,4-二甲氧基化合物37(IC50=0.72μM)对平滑肌瘤细胞(SMCs)的抑制活性最强。
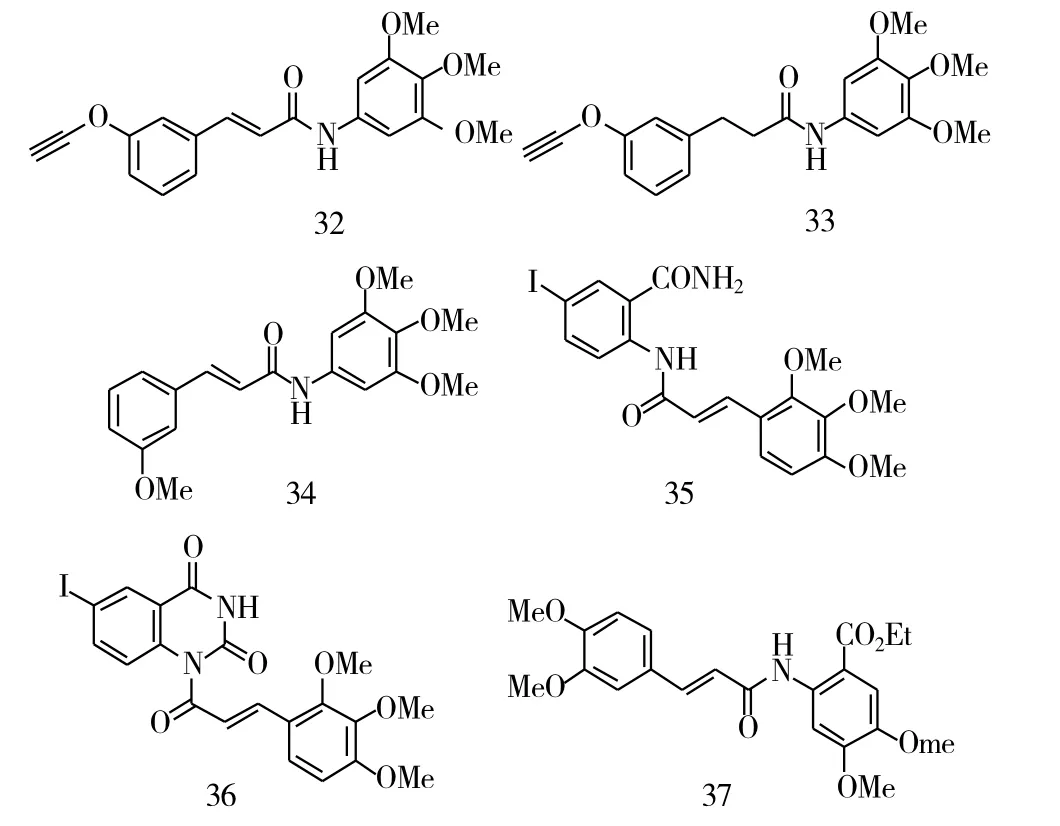
图9 化合物32~37的化学结构
7 其他修饰
在查尔酮类化合物中有一种特殊的二聚查尔酮——姜黄素。姜黄素具有多种药理学活性,包括抗癌活性,因此,可将3,4,5-三甲氧基查尔酮进行二聚,构建出姜黄素类似物。Yadav等[10]构建了一系列3,4,5-三甲氧基的二聚查尔酮,并在α,β-不饱和酮上引入环戊烷、环己烷和哌啶环(化合物38~41,图9),其中含有哌啶环的化合物比含有环戊烷或环己烷的化合物有更强的抗乳腺癌活性,尤其是化合物40,对乳腺癌MDA-MB-231的抑制活性最强,其EC50为0.3μM。但是,进一步将化合物40形成四聚体(化合物42)后,却显著降低了抗肿瘤活性。
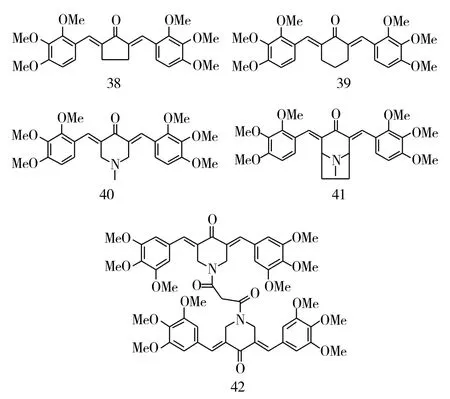
图10 化合物38~42的化学结构
Wei等[11]基于杂环类化合物能够提高抗肿瘤活性的特点,合成了一系列的哌啶、吡喃和噻己环类的化合物。在这些杂环类化合物中,哌啶、吡喃和噻己环类查尔酮 (化合物42~44,图10)的抗肿瘤活性,确实高于环戊环和环己环类的化合物,其中吡喃类化合物的抗肿瘤活性最强,尤其是化合物44,对PC-3、Panc-1和HT-29的抑制活性IC50分别是0.27 μM、0.52 μM 和 0.16 μM。
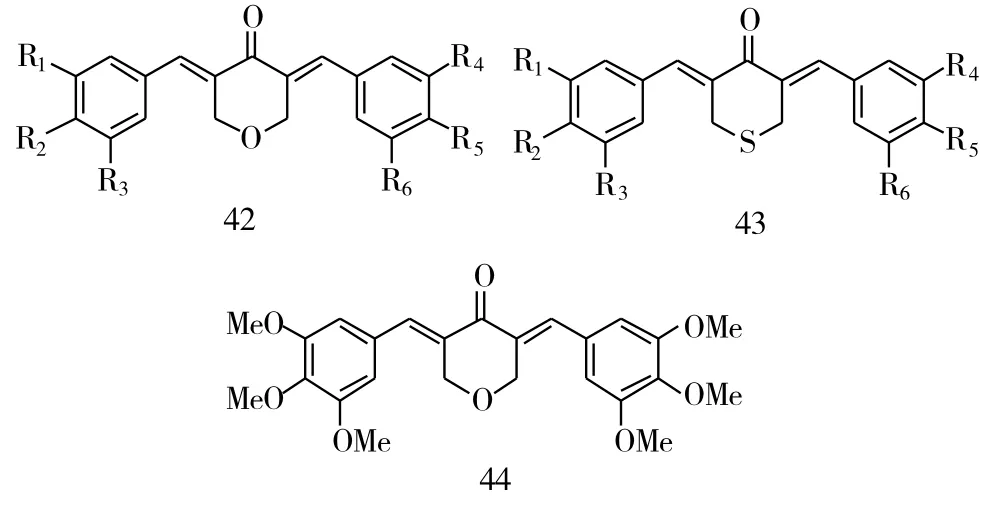
图11 化合物42~44的化学结构
8 结论
本文对近年来3,4,5-三甲氧基查尔酮类化合物的结构修饰研究进行总结,并初步阐述了其构效关系,大致可以总结为以下几个规律:1)3,4,5-三甲基是活性的药效团,改变其甲氧基位置或者减少甲氧基的数量,均会减弱其抗肿瘤活性;2)3,4,5-三甲氧基查尔酮C4亚甲基上引入适当的取代基如甲基,能够提高抗肿瘤活性;3)在3,4,5-三甲氧基查尔酮结构中引入酰胺键,能够提高抗肿瘤活性;4)3,4,5-三甲氧基查尔酮形成二聚体后能够提高抗肿瘤活性。
总之,对3,4,5-三甲氧基查尔酮进行结构修饰能够提高抗肿瘤活性,但是其抗肿瘤效果与临床上应用的药物相比仍然较弱,因此,进一步对3,4,5-三甲氧基查尔酮进行结构修饰是一项非常有前景的工作。
[1] 杨欢,王栋,童丽,等.镰形棘豆的化学成分研究(Ⅰ)[J].中国药学杂志,2008,43(5):338-340.
[2] 郑洪伟,牛新文,朱军,等.查尔酮类化合物生物活性研究进展[J].中国新药杂志,2007,16(18):1445-1449.
[3] Rao Y K, Fang S H, Tzeng Y M. Differential effects of synthesized 2-oxygenated chalcone derivatives: modulation of human cell cycle phase distribution[J]. Bioorg Med Chem.,2004(12): 2679-2686.
[4] Shenvi S, Kumar K, Hattit K S, et al. Synthesis, anticancer and antioxidant activities of 2,4,5-trimethoxy chalcones and analogues from asaronaldehyde: Structure-activity relationship[J]. Eur J Med Chem., 2013, 62: 435-442.
[5] Ducki S, Rennison D, Woo M, et al. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1:synthesis and biological evaluation of antivascular activity [J].Bioorg Med Chem., 2009, 17: 7698-7710.
[6] Lee L, Davis R, Vanderham J, et al. 1,2,3,4-Tetrahydro-2-thioxopyrimidine analogs of combretastatin-A4[J]. Eur J Med Chem., 2008, 43: 2011-2015.
[7] Saxena H O, Faridi U, Kumar J K, et al. Synthesis of chalcone derivatives on steroidal framework and their anticancer activities[J]. Steroids, 2007, 72: 892-900.
[8] Liou J P, Chang Y L, Kuo F M, et al. Concise Synthesis and Structure-Activity Relationships of Combretastatin A-4 Analogues, 1-Aroylindoles and 3-Aroylindoles, as Novel Classes of Potent Antitubulin Agents[J]. J Med Chem., 2004,47: 4247-4257.
[9] Kamal A, Mallareddy A, Suresh P, et al. Synthesis and anticancer activity of 4β-alkylamidochalcone and 4β-cinnamido linked podophyllotoxins as apoptotic inducing agents[J]. EurJ Med Chem., 2013, 63: 501-510.
[10] Yadav B, Taurin S, Rosengren R J, et al. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin[J]. Bioorg Med Chem., 2010, 18: 6701-6707.
[11] Wei X, Du Z Y, Zheng X, et al. Synthesis and evaluation of curcumin-related compounds for anticancer activity[J]. Eur J Med Chem., 2012, 53: 235-245.
Study Advances of Structural Modification and Antitumor Activities of 3,4,5-Trimethoxy Chalcones
HAN Xiao
(Changjiang Polytechnic, Wuhan 430074, China)
Chalcone was a kind of natural polyphenol widely found in natural plants and had various biological activities included anti-tumor, anti-bacterial, antiviral activities. Recently, a great deal of structural modif i cations on chalcone have found 3,4,5-trimethoxy chalcones was a new kind of chalcone with good anti-tumor activity. In this paper, the research advances of structural modif i cation of 3,4,5-trimethoxy chalcones, as well as anti-canceractivities were reviewed, and those would provided assistances for the future development of 3,4,5-trimethoxy chalcone drugswith high and specif i c anti-tumor activity.
3,4,5-trimethoxy chalcones; structural modif i cation; anti-canceractivity
R 284.3
A
1671-9905(2017)10-0029-05
湖北省教育厅科学技术研究项目(B2017578)
韩潇(1984-),男,湖北宜昌人,硕士,长江职业学院生物医药学院讲师,研究方向为生物制药技术。E-mail:hxiao12378@126.com
2017-07-10
